
A heating-up method for the synthesis of pure phase kesterite Cu2ZnSnS4 nanocrystals using a simple coordinating sulphur precursor†
Abstract
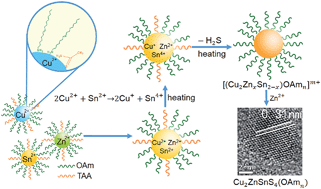
Pure phase kesterite Cu2ZnSnS4 (CZTS) nanocrystals (NCs) have been successfully synthesized via a heating-up method utilizing metal salts as cation sources and thioacetamide (TAA) as a coordinating sulphur precursor in combination with oleylamine (OAm) as a coordinating ligand and solvent. The slower release of H2S from the metal–TAA complexes, as compared with the commonly used sulphur powder, is crucial for control of the grain size and size distribution of the NCs. As dominating coordinating ligands, OAm led to Sn-rich nuclei and kesterite CZTS NCs. The formation of pure phase kesterite CZTS NCs depends significantly on the temperature and Zn2+/Sn4+ molar ratio of the reaction system. The formation mechanism of the pure phase kesterite CZTS NCs has been clarified taking into account the chemical potentials of the elements involved.
 Please wait while we load your content...
Please wait while we load your content...

