
Interactions in globular proteins with polyampholyte: coacervation route for protein separation†
Jyotsana Pathaka,
Kamla Rawat*bc,
V. K. Aswald and
H. B. Bohidarab
aPolymer and Biophysics Laboratory, School of Physical Sciences, Jawaharlal Nehru University, New Delhi 110067, India
bSpecial Center for Nanosciences, Jawaharlal Nehru University, New Delhi 110067, India. E-mail: bohi0700@mail.jnu.ac.in; kamla.jnu@gmail.com; Fax: +91 11 2674 1837; Tel: +91 11 2670 4699
cInter University Accelerator Centre (IUAC), New Delhi 110067, India
dSolid State Physics Division, Bhabha Atomic Research Centre, Mumbai-400085, India
First published on 19th January 2015
Abstract
In this work, we report exclusive separation of Bovine Serum Albumin (BSA) from a solution where this protein was present with β-lactoglobulin (β-Lg) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.75 (w/v) ratio at their common isoelectric pH (5 ± 0.02). A polyampholytic polypeptide Gelatin B (GB) also having the same pI was used to extract protein (BSA or β-Lg) molecules selectively from this solution through a process called complex coacervation. In our study, the protein-rich condensate, called coacervate, comprised of GB–BSA complexes while the supernatant mostly contained β-Lg molecules. For the separation of BSA from BSA–GB coacervate, we used ethyl alcohol, which removed the BSA to the supernatant. The differential binding affinity of BSA versus β-Lg to GB chains was established through fluorescence quenching and fluorescence resonance energy transfer (FRET) studies. The BSA–GB binding protocol followed a surface selective patch binding mechanism and these results were obtained from an array of experimental methods such as UV-vis and fluorescence spectroscopy, small angle neutron scattering (SANS), FTIR and circular dichroism spectroscopy. Herein, it is clearly established that selective coacervation at pI can be used as a method for protein separation.
:0.75 (w/v) ratio at their common isoelectric pH (5 ± 0.02). A polyampholytic polypeptide Gelatin B (GB) also having the same pI was used to extract protein (BSA or β-Lg) molecules selectively from this solution through a process called complex coacervation. In our study, the protein-rich condensate, called coacervate, comprised of GB–BSA complexes while the supernatant mostly contained β-Lg molecules. For the separation of BSA from BSA–GB coacervate, we used ethyl alcohol, which removed the BSA to the supernatant. The differential binding affinity of BSA versus β-Lg to GB chains was established through fluorescence quenching and fluorescence resonance energy transfer (FRET) studies. The BSA–GB binding protocol followed a surface selective patch binding mechanism and these results were obtained from an array of experimental methods such as UV-vis and fluorescence spectroscopy, small angle neutron scattering (SANS), FTIR and circular dichroism spectroscopy. Herein, it is clearly established that selective coacervation at pI can be used as a method for protein separation.
1. Introduction
In the early days of protein chemistry, the only practical way to separate different types of proteins in industry was by chromatography, electrophoresis, precipitation, centrifugation and membrane separation. The many forms of these techniques offer high resolving power and good selectivity but are limited by low throughputs, large solvent and time consumption and expense.1,2 Membrane chromatography has very good characteristics for biomolecular purification.3 It is easy to scale up and set up. However, the quality of these membranes, for example stability of coating and evenness of thickness, do affect the efficacy of separation. So there is a need for large scale, economic and highly selective technique as alternative to these techniques for protein purification. The first use of polyelectrolyte complexation coacervation for protein separation may be dated back to the early 50's, when Morawetz and Hughes4 proposed that proteins could be isolated from a binary mixture by the addition of a polyelectrolyte. They demonstrated that BSA and oxyhemoglobin could be separated in the inter-isoelectric range by polymethacrylic acid. A similar separation scheme was later applied to many other systems.5–20 Sternberg and Hershberger7 using polyacrylic acid (PAA) selectively separated a mixture of equal amounts of lysozyme, (3-galactosidase, a-amylase, and protease) into different fractions, which were enriched in different proteins. Shieh and Glatz also used polyacrylic acid (PPA) to separate lysozyme from ovalbumin.8 Xia and Dubin summarized the use of polyelectrolytes for protein separations and showed that over 90% recovery was achieved for most systems.9 After that, the separation of protein via polyelectrolyte was reported by Xu et al., in which BSA/β-Lg was separated by cationic polyelectrolyte poly(diallyldimethylammonium chloride) (PDADMAC).20The phenomenon of complex coacervation is ubiquitous both in nature and in man-made materials like the natural underwater glue of the sandcastle worm,11 the bacterial nucleoid and the pectin-coated casein in the yogurt drinks.12–14 Colloidal systems containing a mixture of positive and negatively charged macromolecules in water often form coacervates. DNA is compacted by positive charged histone proteins into complex coacervates that fold into chromatin fibers at cellular level.15–18 In blood clotting mechanism the negatively charged phospholipids combine with positively charged calcium-rich GLA domain of clotting proteins.19 Caddisfly larvae build composite retreats out on the rocks and sticks using underwater silk protein complex using the similar strategy.20–22 Protein and polysaccharide complexation and coacervation have been widely used to confer structure, stabilize food products and to provide desired functionality.23–26 Practical applications such as microencapsulation27 of active ingredients, protein separation28,29 and purification30 in the pharmaceutical industry also utilize the technique of intermolecular complexation and coacervation.31–33
A pertinent question arises here: how to separate a pair of protein molecules having a common pI, and similar zeta-potential vs. pH profile? We have comprehensively answered this question herein. Solubility of proteins close to their iso-electric pH is minimum because the protein net charge at pI is zero. However, protein molecules are associated with heterogeneous charge distribution throughout the pH range. Therefore, protein–protein interaction is strongly pH dependent. We have exploited this property and have successfully separated Bovine Serum Albumin (BSA) from a solution where this protein was present with β-lactoglobulin (β-Lg) in 1:0.75 ratio (w/v) at their common pI (5 ± 0.02) through the phenomenon of complex coacervation. This work aims to establish that selective coacervation at pI can be utilized as an alternative method for protein separation. The advantages of this separation technique are: (i) simplicity with respect to instrument (ii) low cost (iii) solvent economy, and (iv) reasonably high speed.
2. Material and methods
Gelatin B (bovine skin extract, bloom = 225 and molecular weight 100 kDa), β-Lg (molecular weight 18 kDa) and BSA (molecular weight 67 kDa) were bought from Sigma-Aldrich chemical company (U.S.A.), which according to supplier had a minimum purity of >98%. It needs to be mentioned that the proteins were not subjected to further purification by dialysis, which would have made these salt free. GB, β-Lg and BSA samples were used as received. All concentrations mentioned are in the units of (w/v) unless otherwise stated.Turbidity is an indirect measure of the binding between the protein/polyampholyte and its magnitude is proportional to the concentration and geometrical shape and size of the products formed. The change in transmittance (%T) of the solution was monitored continuously using a turbidity meter (Brinkmann-910, Brinkmann Instruments, USA) operating at 450 nm using a 1 cm path length probe, and it was calibrated to 100% transmittance with deionised water.
The transmittance and pH change of the mixture were noted throughout by titrating with 0.1 N NaOH or 0.1 N HCl as required with gentle magnetic stirring. Solution turbidity is given by (100 − %T) and the fluctuations (±0.1%) of transmittance were treated by consistently selecting the highest transmittance value.
FT-IR spectra from all samples were recorded on a FT-IR/Raman Spectrometer (1064 nm) attached with a Microscope (Varian 7000 FT-Raman and Varian 600 UMA). The steady state fluorescence measurement was performed using Varian Cary eclipse fluorescence spectrophotometer with spectral range 190 nm to 1000 nm, using 5 nm slit width. A 1 cm path length rectangular quartz cell was used as sample holder for these studies. Also, appropriate blank corresponding to the buffer was subtracted to correct for the fluorescence background. The experiments were repeated and found to be reproducible within experimental error.
Circular dichroism (CD) experiments were carried out with Applied Photo physics Chirascan instrument (USA) to estimate the secondary structure of proteins using the standard operation procedure. Each spectrum was the average of three successive scans. Appropriate baseline corrections in the CD spectra were made. The path length of the cuvette used in the CD experiments was 0.1 cm and the wavelength range used was from 200 to 260 nm. It has been reported that below ∼200 nm CD data are not very accurate for analysis of protein secondary structure.34
Small angle neutron scattering (SANS) requires a neutron source, i.e., a nuclear reactor or an accelerator-based spallation source, and therefore, the experiments are performed at large scale facilities. The small angle neutron scattering experiments presented in this work were performed at the SANS diffractometer at the Guide Tube Laboratory, Dhruva Reactor, Bhabha Atomic Research Centre, India. It makes use of polycrystalline block of beryllium oxide (BeO) filter as monochromator. The mean wavelength of the monochromatic beam was 5.2 Å with a spread of Δλ/λ ∼ 15%. The angular distribution of neutrons scattered by the sample was recorded using a 1 m long one-dimensional He position sensitive detector. The instrument covered a q-range of 0.015–0.35 Å−1. The coacervate samples were transferred to a quartz cell having a thickness of 2 mm, and scattered neutron intensity I(q) was measured as a function of scattering vector, q. The measured intensity was corrected for the background and for the empty cell contribution, and the data were normalized to get the structure factors.35
BSA and β-Lg aqueous solutions were prepared by dissolving known amount of the protein powder in double distilled deionized water at 25 °C using a magnetic stirrer for almost 1 hour. GB aqueous solution was prepared by dissolving known amount of the protein powder in double distilled deionized water at 40 °C using a magnetic stirrer for almost 1 to 1.5 hours. These stock solutions appeared optically clear and transparent to the eye. All procedures were performed at room temperature 25 °C and relative humidity in the laboratory was less than 50%. The mixing ratios of the two proteins (BSA–GB) and (β-Lg–GB) was varied in the range, r = 0–2 (r = BSA:GB or β-Lg:GB).
For the preparation of mixed solutions of BSA/β-Lg/GB, we dissolved BSA, β-Lg and GB for 1 hour in double distilled deionized water using a magnetic stirrer to give 10 g L−1 BSA, 10 g L−1 GB and 7.5 g L−1 β-Lg stock solutions. For observing separation of BSA and β-Lg as a consequence of binding to GB, solution of BSA, β-Lg and GB were all mixed together (same volume) and pH was adjusted to 5.0 ± 0.2. To initiate coacervation, this solution was titrated with 0.1 N HCl and 0.1 N NaOH until turbidity maxima was found.20–22
A small amount of sodium azide (1 mg L−1) was added to these turbid samples to prevent bacterial contamination. These were stored in air tight borosilicate glass bottles for 10 days. Coacervates were extracted from reacted solutions following standard procedure of repeated centrifugation and decantation of the supernatant.31,36–39 This was repeated at least three times, which yielded the coacervates. Coacervate samples prepared in D2O were used for SANS experiments. The absorbance (UV-vis spectrophotometer, Model CE-7300, Cecil Instruments, UK) of the solutions were measured at λ ≈ 290 nm (maximum BSA, β-Lg absorption wavelength).
3. Results and discussion
The experiments were performed in two distinct steps. First the differential binding between the two proteins and GB was evaluated. In the second step, coacervation phenomenon was used as a protocol for protein separation. The BSA and β-Lg molecules are associated with nearly spherical conformation and have hydrodynamic radius Rh = 3.1 and 3.4 nm respectively.40 GB is a random coil biopolymer with radius of gyration, Rg = 34 nm and hydrodynamic radius, Rh = 23 nm. This yields Rh/Rg ≈ 0.67 implying GB molecule has a random coil conformation.41 Further, GB is associated with a persistence length of 2 nm.42 The heterogeneous charge distribution in the three molecules enable surface patch binding mediated associative interaction.433.1 Differential binding behavior of BSA/β-Lg to GB
The UV-vis absorption spectra of proteins (BSA, HSA, β-Lg) showed absorption bands located at ≈280–290 nm, reflecting the presence of tryptophan (Trp) and tyrosine (Tyr) residues. In all the proteins there was a strong absorption band at around 280–290 nm which was mainly due to the transition of π–π* of protein's characteristic polypeptide backbone structure and the aromatic ring portion of their structure. The UV-vis absorbance from the protein mixture solutions were noted by varying the protein concentration (BSA–β-Lg) but keeping the polyampholyte concentration fixed as shown in Fig. 1. Absorption of GB is characterized by a strong band in the UV region at 290 nm. Addition of BSA or β-Lg led to gradual increase in GB absorption. These observations indicate that there is a structural change (microenvironment) in GB which has occurred upon interaction with the BSA or β-Lg. The baseline correction was done for all UV spectral measurements. So we confirmed that the increase in optical density is only resulting from the interaction between GB and BSA or β-Lg not due to scattering. After a certain ratio the absorbance become almost constant in all BSA–GB, β-Lg–GB and BSA–GB–β-Lg pair i.e. there in no structural change at those ratios. This ratio (r) is taken as maximum binding condition in all pairs. The concentration of GB at r = 0 is 1% w/v, while the concentration of other protein is varying. At r = 1 almost all GB molecule interacted with BSA or β-Lg, after that at r = 1.5 slightly increase absorbance is due to excess BSA or β-Lg. The binding plot indicated that the optimum binding between BSA–GB and β-Lg–GB occurred at the mixing ratio 1:1 and 0.75:1 respectively. This preset the condition for other experiments.
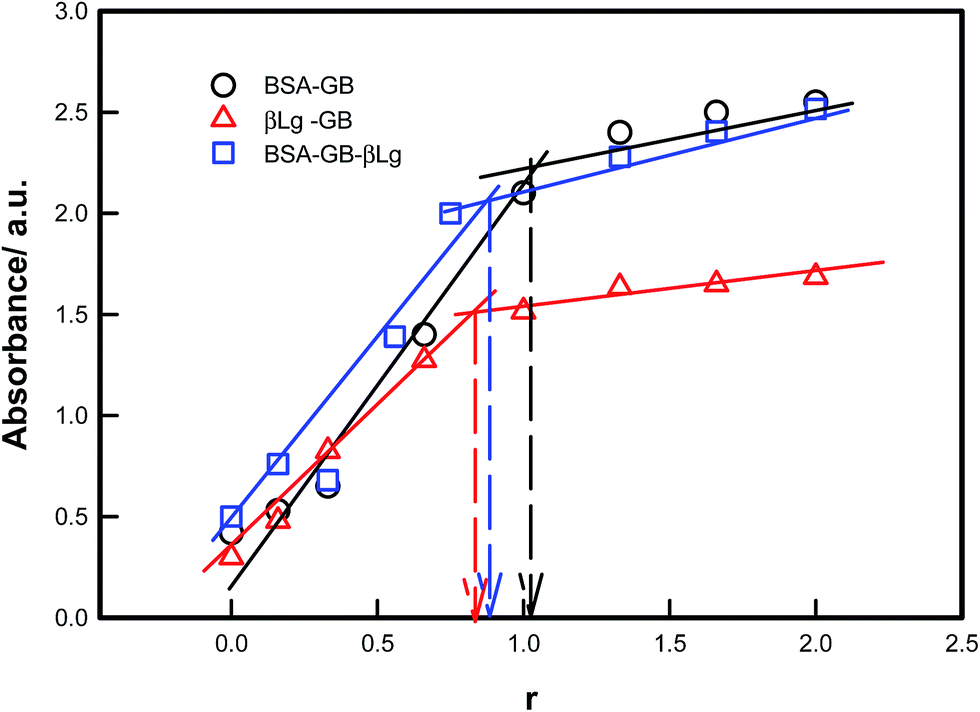 | ||
| Fig. 1 Binding plot of BSA/β-Lg with gelatin B and BSA–GB with β-Lg measured at 25 °C which clearly shows the optimum binding condition pertained to r = BSA/GB = 1, β-Lg/GB = 0.75 and BSA–GB/β-Lg = 0.75 (arrows). | ||
The turbidity titration of the solution containing the two proteins in the different volumetric mixing ratio was performed by slow addition of NaOH and the solution turbidity was monitored to determine the critical pH responsible for phase separation (Fig. 2). Three signature pHs defined coacervation transition: (i) formation of soluble complexes at pHc, sharp change in turbidity, (ii) coalescence of soluble complexes and onset of coacervation transition, pHϕ, turbidity maximum and (iii) formation of large insoluble complexes at pHprep, noticed as reduction in the turbidity value.
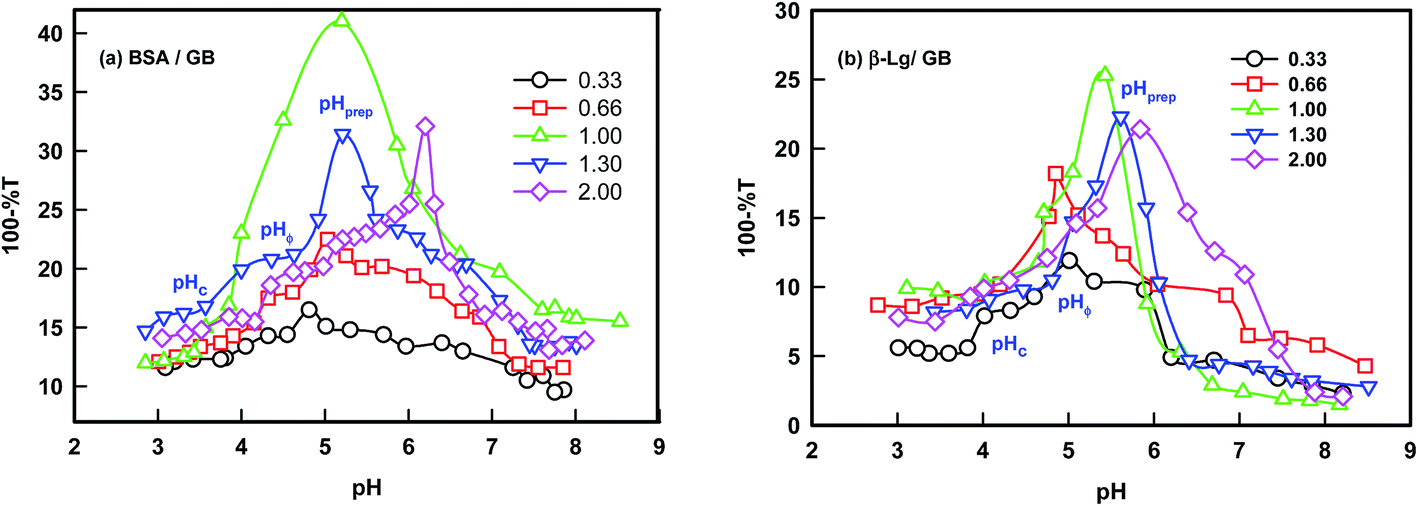 | ||
| Fig. 2 Determination of coacervation transition of BSA/β-Lg with GB at various stoichiometric binding ratios. Typical signature phase separation pHs (pHc, pHϕ and pHprep) are shown on one of the curves. | ||
Turbidity was seen to increase with pH (Fig. 2) which was attributed to the formation of soluble complexes due to the interaction between cationic amine groups of the BSA or β-Lg molecules and anionic carboxylate groups of the GB chain.
The solubility of the complexes was found to be dependent on the (BSA–GB) or (β-Lg–GB) mixing ratio, the degree of ionization of the GB carboxyl and amine groups, and the same of the BSA/β-Lg molecules. At higher mixing ratio (r ≥ 1), the pH at which turbidity maxima was observed was at pH = 6, reflecting the delicate interplay of the charge on protein/polyampholyte molecule and mixing ratio. The pH at which the turbidity remained constant at higher r indicated that the maximum binding had been achieved. The effect of mass ratio of protein/polyampholyte was found to depend on the critical pH values in both cases. Data shown in Fig. 1 clearly indicated that the optimum stoichiometric binding condition for the two proteins was: r = [BSA]:[GB] = 1 and [β-Lg]:[GB] = 0.75.
The simplicity and sensitivity of turbidimitric titration method as applied to protein–polyampholyte systems is based on the fact that turbidity is proportional to both the molecular weight and the number density of particles present in dispersion. The change in turbidity mirrors the extent of interactions between the two biopolymers (gelatin and BSA or β-Lg) prevailing at an instance. Typically, a mixed solution was kept on a magnetic stirrer, and was stirred at moderate speed with stir bars. Such a solution was titrated with 0.1 M NaOH and the transmittance and pH changes of the mixture were noted throughout. We observed the first occurrence of turbidity corresponding to the formation of soluble complexes at pHc. The titration process was continued until maximum turbidity (pHϕ) was noticed. The titration profiles are shown in Fig. 2 for various mixing ratio of gelatin and other two proteins. These transition pHs are well defined and discussed, in general, for coacervating systems in the past for complex coacervation.21–31 The soluble complexes could be formed in a very narrow range of pH. At pHc, the initiation of intermolecular soluble aggregate formation comprising charge neutralized protein–gelatin complexes ensued. Eventually, these led to the formation of microscopic coacervate droplets which in turn coalesce through Ostwald ripening to minimize the surface free-energy and macroscopic droplets were generated. In this process, the growth of larger droplets at the expense of smaller ones is facilitated. Normally, for pH > pHϕ one observes the formation of large insoluble complexes that undergo precipitation immediately, which is observed in the turbidity-pH profile as a sharp drop in measured turbidity value (pHprep).
The specific pH regions of aggregation between BSA/β-Lg with GB can be utilized for setting up a protein separation and purification protocol. Both the proteins (BSA and β-Lg) have comparable size (hydrodynamic radius, 3.1 and 3.4 nm for BSA and β-Lg respectively) and nearly same isoelectric pH (pI = 4.9 and 5.1 to BSA and β-Lg respectively). The optimum binding between protein and polyampholyte could be generated by bulk stoichiometry while the separation efficiency could be tuned by changing the modulating pH. The turbidity titration data of BSA/GB and β-Lg/GB are shown in Fig. 2 which was used to determine the pHs of soluble complex formation (pHc), and liquid–liquid phase separation (pHϕ). Direct comparison of data presented in Fig. 2 shows that the onset of binding for BSA and β-Lg with GB was not identical. Results shown in (Fig. S1, ESI†) indicated that pHc was higher for BSA at all binding ratio meaning that it was bound to GB more strongly in comparison to β-Lg. A phase diagram was constructed from the turbidity data to map the binding of BSA/β-Lg with GB as function of mixing ratio. The phase boundary could be observed clearly (Fig. S1, ESI†).
Fig. 3 shows the turbidity varying with pH for the interacting biopolymer pairs. The shaded regions display the stable zone for intermolecular soluble complex formation. Thus, the shaded region qualitatively represents the typical yield of coacervation (area of the shaded region) and the corresponding values are plotted,43 which is shown as inset of Fig. 3. This shows that strong associative interaction prevail in BSA–GB system, leading to large coacervation yield. The coacervation yield is depicted as bar diagram which implies BSA–GB yield was 3 times more than the β-Lg–GB case. In order to determine the coacervation yield as a function of β-Lg concentration, a series of pH titrations of BSA–GB–β-Lg solutions were carried out. It was found that maximum coacervation yield pertained to the β-Lg concentration ≈0.75% (Fig. S2, ESI†).
 | ||
| Fig. 3 The pH dependent turbidity profile of the interacting solution containing BSA–β-Lg with GB, and BSA with β-Lg. Shaded region shows the coacervation domain for various pairs. Inset shows the coacervation yield. | ||
3.2 Fluorescence spectra and quenching
Fluorescence spectroscopy is useful to obtain local information about the conformational and dynamic changes occurring in proteins. In proteins, the intrinsic fluorescence is high due to the presence of Tryptophan and it is also sensitive to the local environment. Changes in the fluorescence emission spectra of protein often occur in response to conformational transitions, subunit association, substrate binding, or denaturation. In BSA the tryptophan residue involved in binding could be either Trp-134 or Trp-212. Trp-134 located on the surface of albumin molecule, is more exposed to hydrophilic environment, whereas Trp-212 is deeply buried inside the hydrophobic pocket of the protein.44 β-Lg has a 2 tryptophan residues, Trp-19 and Trp-16. Thus, on observing the fluorescence emission of Trp in the bio-conjugates, information about the protein conformational behavior around the Trp residues can be obtained. Gelatin contains ca. 1% tyrosine, 2–3% phenylalanine, and no tryptophan.45 Phenylalanine is not excited in most of the cases and its quantum yield is rather low, so the emission from this residue can be ignored. Hence, the fluorescence exclusively results from tyrosine in the gelatin.Fig. 4 shows the emission spectra of BSA and β-Lg in absence and presence of GB excitation at 295 nm. As shown the intensity decreased in BSA/GB with addition of GB while in β-Lg–GB case intensity did not change for peak located at 340 nm. In BSA, Trp fluorescence quenched drastically with the addition of GB, this indicated BSA bound strongly to GB while in case of β-Lg, the Trp fluorescence quenching was very less. Thus, β-Lg was weakly bound to GB. The extent of quenching of intrinsic fluorescence of proteins (at 340 nm) by binding to GB molecules could be described by Stern–Volmer equation given by46
| I0/I = 1 + Kqτ0[GB] = 1 + KSV[GB] | (1) |
 | ||
| Fig. 4 Steady state fluorescence spectra (λexc = 295 nm) of (a) BSA and (b) β-Lg systems with addition of GB. | ||
The binding constant, K and number of active binding sites, n between BSA/β-Lg and GB were calculated from eqn (2) from the quenching data.47–49
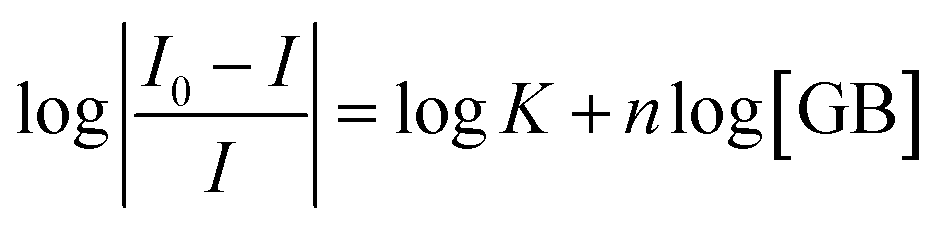 | (2) |
A plot of log[(I0 − I)/I] vs. log[GB] gives a straight line (Fig. 5), whose slope equals to n and the intercept on Y-axis equals to logK. The least-squares fitting values are listed in Table 1.
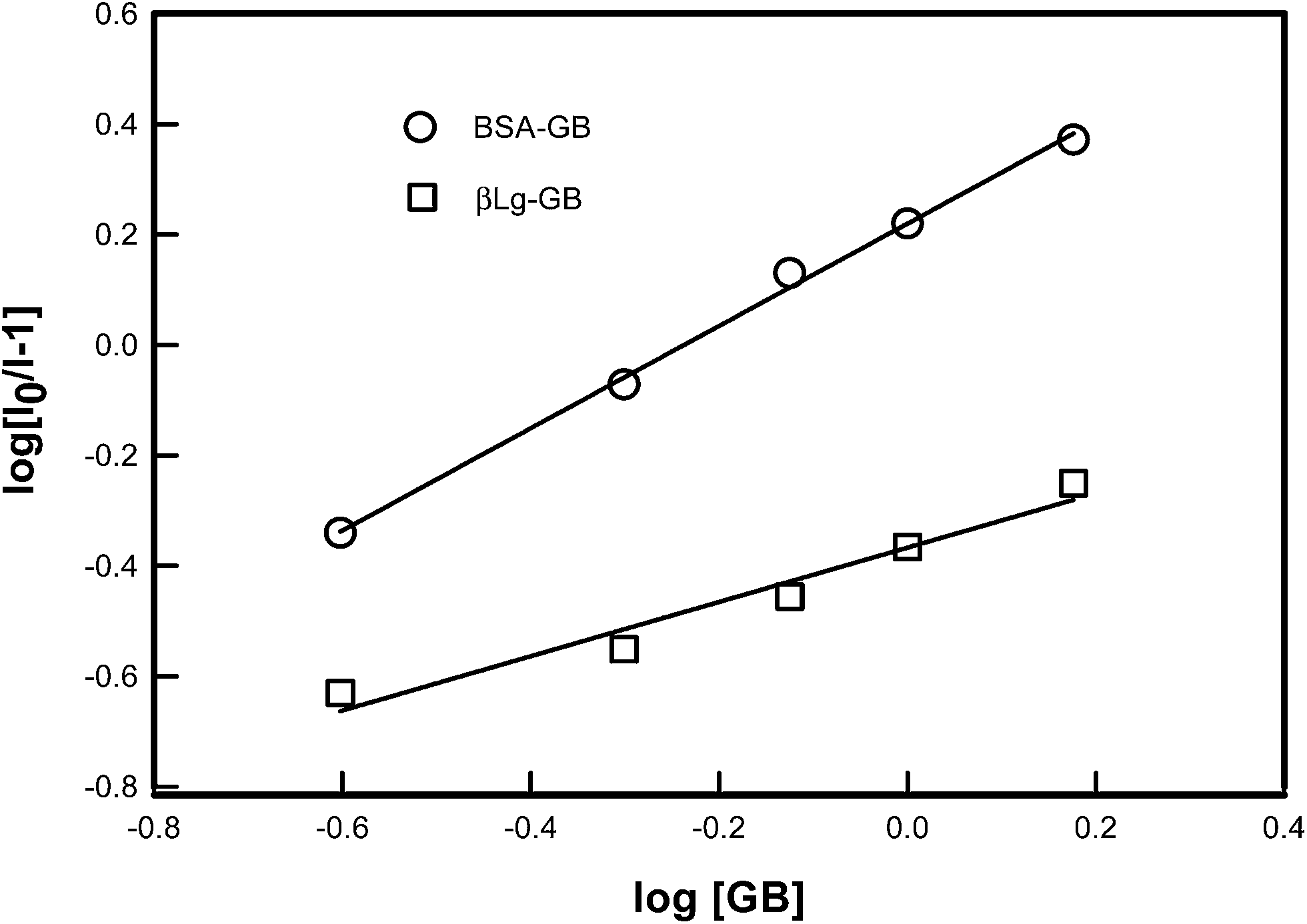 | ||
| Fig. 5 Logarithmic plot derived from fluorescence data of various proteins [BSA, β-Lg] as a function of concentration of GB. The binding constant K and number of binding sites n was determined from the intercept and slope of least square fitted straight line to the data points as described by eqn (2). | ||
| Samples | K/M−1 | n | Kq/M−1 S−1 |
|---|---|---|---|
| a From the fluorescence spectra, the binding constant of BSA–GB was found to be greater than that of β-Lg–GB. Thus, we conclude that GB was bound more strongly to BSA compared to β-Lg. | |||
| BSA–GB | 162 ± 0.15 | 1 | (43.0 ± 0.5) × 1010 |
| β-Lg–GB | 43 ± 0.01 | 0.50 | (1.4 ± 0.2) × 1010 |
3.3 Energy transfer and binding distance
The FRET theory is a non destructive spectroscopy method that can be used to determine the mean distance between amino acid residues (the donor) and bound molecules (the acceptor). There is a good overlapping between the fluorescence emission spectrum of free BSA/β-Lg and absorption UV spectra of GB (Fig. 6). As the fluorescence emission of protein was affected by the excitation wavelength, λexc = 295 nm, the spectrum ranging from 250 to 500 nm was chosen to calculate the overlapping integral.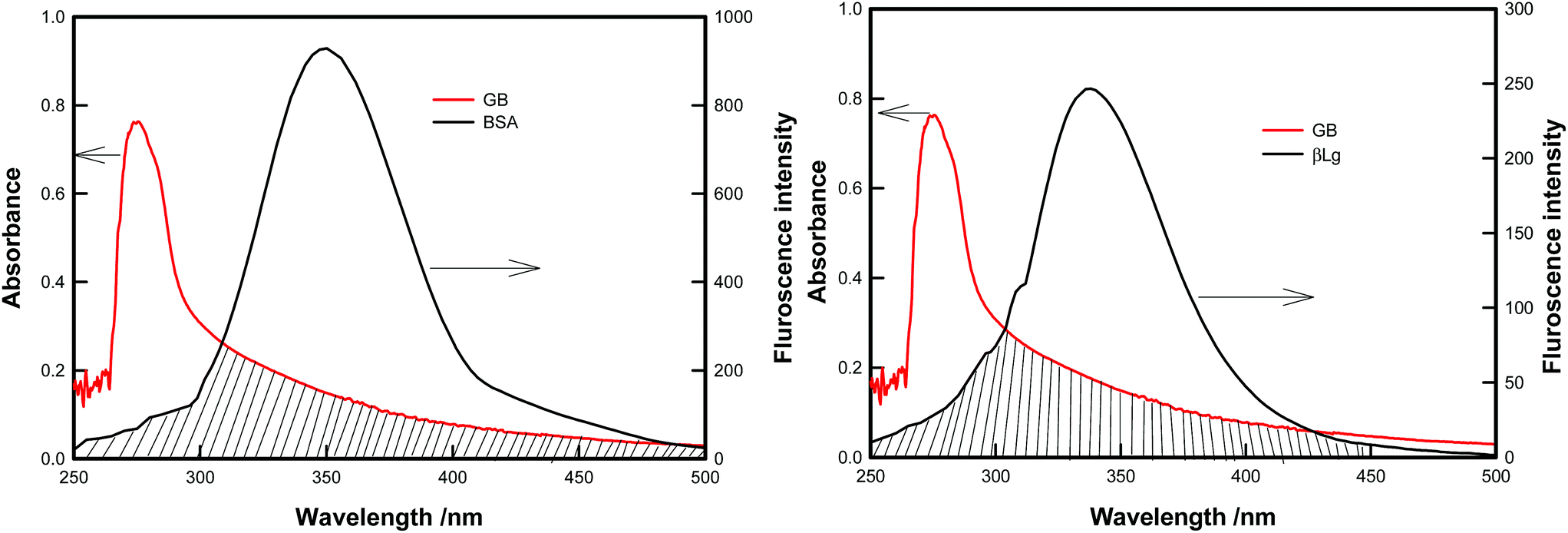 | ||
| Fig. 6 Overlap spectra between the fluorescence emission of BSA/β-Lg and UV absorption of GB. | ||
Förster (or fluorescence) resonance energy transfer (FRET) between two molecules is typically observed over distances of less than 10 nm. This is because this process results from dipole–dipole interactions, and hence depends on center-to-center separation between the donor and acceptor molecules. In particular, it necessitates a finite overlap between donor emission and acceptor absorption spectra.50 FRET efficiency is strongly dependent on the distance separating the FRET pair and the relative orientation of the molecules. FRET occurs between a donor molecule in the excited state and an acceptor on the ground state. The donor molecule emits shorter wavelength emission spectrum that overlaps with the absorption spectrum of acceptor, and result in the long range dipole–dipole interaction between the donor and the acceptor pair. In the study of proteins the donor is tryptophan residue, so the FRET mechanism allowed for the determination of distance between Trp residue of protein (BSA/β-Lg) and the GB.
The FRET efficiency, E of donor–acceptor pair separated by distance r can be expressed by using the Forster formula given as51,52
 | (3) |
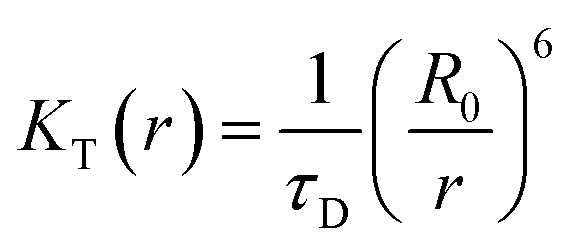 | (4) |
| R06 = 8.79 × 10−25K2n−4ϕJ(λ) | (5) |
K2 is the spatial orientation factor, which describes the relative position of the donor and acceptor dipoles.53,54 Ranging from 0 (perpendicular dipole) to 4 (parallel dipole). generally, the dipoles are assumed to be rapidly moving, on time scale similar to the donor excited-state lifetime, and their orientation are therefore described as random, with K2 = 2/3; n is the refractive index of medium; ϕ is the quantum yield of the donor in the absence of acceptor; J expresses the degree of spectral overlap between the donor emission spectrum and the acceptor absorption spectrum and the UV absorption spectrum of the acceptor and was calculated by dividing the area of the overlapped region to a very small rectangle, which could be calculated from the following equation53,54
| J(λ) = ∫FD(λ)εA(λ)λ4dλ | (6) |
In BSA the tryptophan residue involved in binding could be either Trp-134 or Trp-212. Trp-134 located on the surface of albumin molecule, is more exposed to hydrophilic environment, whereas Trp-212 is deeply buried inside the hydrophobic pocket of the protein. β-Lg has a 2 tryptophan residues, Trp-19 and Trp-16. The value of r and R0 are less than 10 nm, and fulfil the required condition 0.5 R0 < r < 1.5 R0 indicating that the energy transfer from tryptophan residue to GB was a possibility. Table 2 summarizes the FRET parameters relevant for our system. In BSA larger E value and the smaller r value indicates high energy transfer efficiency, while in β-Lg low E value and the larger r value indicates low energy transfer efficiency, which is consistent with the larger binding constant (K) associated with BSA–GB interaction (Table 1).
| Protein | J(λ) × 1011/cm3 M−1 | R0/nm | r/nm | E/% |
|---|---|---|---|---|
| β-Lg | 2.4 | 8.3 | 9.2 | 30 |
| BSA | 3.1 | 4.8 | 4.3 | 63 |
3.4 Effect of binding on secondary structure
The basis of CD spectroscopy is the difference of absorption of right-handed and left-handed circularly polarized light passing through a sample and radiation by chromophores which either possess intrinsic chirality or are placed in chiral environments. This difference spectrum is related to chilarity of the proteins. Proteins possess a number of chromophores which can give rise to CD signals. In the far UV region (250–180 nm), which corresponds to peptide bond absorption, the CD spectrum can be analyzed to give the content of regular secondary structure features such as α-helix and β-sheet. Circular dichroism spectroscopy is a quantitative technique to investigate the secondary structure of proteins in aqueous solution. Integrity of secondary structure has bearing on the biological activity of proteins.The CD spectra of proteins and their GB bound complexes are shown in Fig. S3 (ESI†) at pH = 5 ± 0.2. All mix samples of BSA–GB and β-Lg–GB were turbid and this resulted in the poor CD signal. Therefore these samples were diluted to very low concentrations.
It was observed that when GB was added to BSA the characteristic peaks (two negative double humped peaks) of high α-helix content in BSA became deeper whereas for β-Lg case when GB was added the characteristic peak was similar to GB characteristic peak which changed only due to increase in concentration of GB. Since the α-helix is one of the elements of secondary structure, the structure change of albumin then could be evaluated from the content of the α-helix structure (denoted as helicity).
The α-helix content of BSA–GB decreased, which suggested strong interaction between BSA and GB whereas in β-Lg-GB system very weak interactions prevailed. The decreased percentage of α-helix content in BSA structure indicated that GB was bound to the amino acid residues of the main polypeptide chain of proteins and destroyed their hydrogen bonding networks.55
The CD result was expressed in term of mean residue ellipticity (MRE)56 in deg cm2 dmol−1.
 | (7) |
 | (8) |
The CD data was used in eqn (8) to determine the helix content (secondary structure) of various molecular complexes which is plotted in Fig. 7. From CD study it was noticed that increasing concentration of GB, BSA helicity decreased indicating binding of GB to BSA, while in case of β-Lg helicity did not changes very much. The observed change in CD spectra was due to increase in GB concentration which indicated GB was weakly bound to β-Lg.
 | ||
| Fig. 7 Dependence of secondary structure (helicity) of proteins (BSA, β-Lg) as function of binding ratio. | ||
3.5 Separation of BSA and β-Lg
In the second step, we shall be discussing the use of coacervation in protein separation. The three components were mixed in same volume at pH = 5 and the final concentrations were 1% BSA, 1% GB and 0.75% of β-Lg. Whereas the turbidmetric titration, fluorescence spectra and CD spectra showed stronger binding of GB with BSA, the extent of separation possible was not evident from these results. From the titration of BSA–GB samples at different weight ratio, and in the presence and absence of β-Lg (Fig. 8(a)), it was seen that β-Lg had no influence on the coacervation of BSA with GB. This indicated that proteins could interact independently with GB. After centrifugation of this solution at pH 5, two layers (bottom and top) were identified as coacervate, and supernatant. The property of the BSA–β-Lg–GB coacervate was compared with BSA–GB coacervate from SANS studies (Fig. 8(b)), and those for supernatant were compared with initial protein solutions by UV-visible and FT-IR analysis (Fig. 9). Fig. 10 shows that BSA–GB titration curve in absence and presence of β-Lg which indicates that β-Lg had no influence on BSA–GB coacervation. Also, the analysis of coacervate of BSA–GB in presence, and absence of β-Lg by SANS studies showed that β-Lg had no influence on BSA–GB coacervation.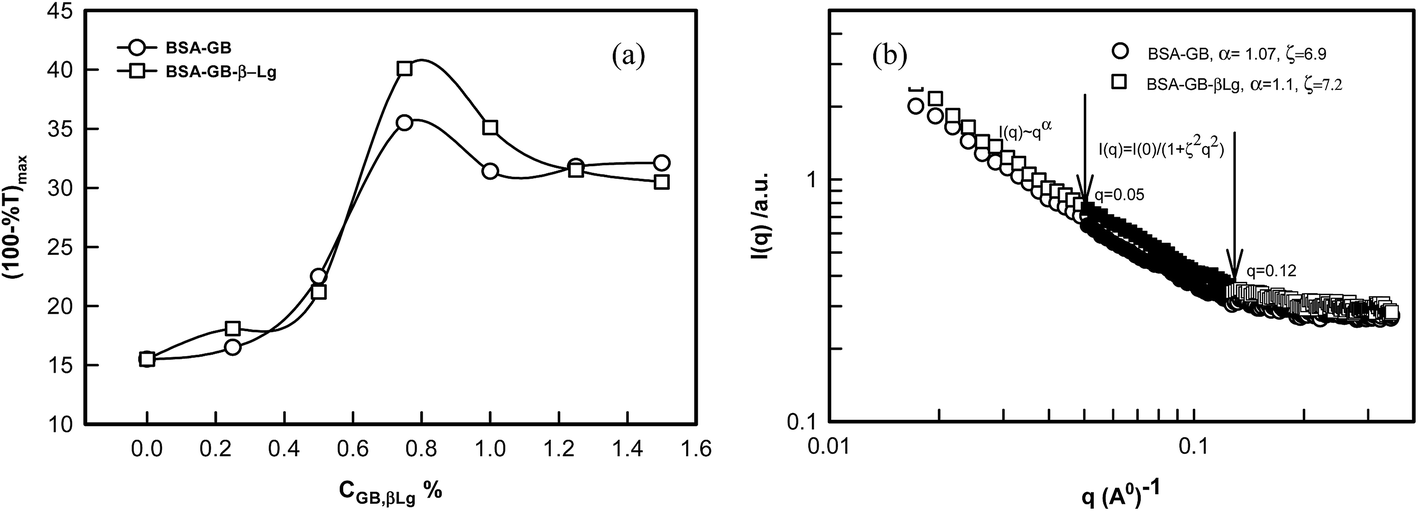 | ||
| Fig. 8 (a)Turbidmetric titration of BSA and GB with and without β-Lg. (b) Neutron scattering intensity profile of a BSA/GB coacervate with and without β-Lg. Arrows indicate the distinctive q-regions where the power-law and Ornstein–Zernike regimes prevailed. See text for details. | ||
 | ||
| Fig. 9 (a) UV-visible spectra of BSA, β-Lg and GB, with BSA/β-Lg/GB supernatant. (b) FTIR spectra of BSA, β-Lg and GB, with BSA/β-Lg/GB supernatant. | ||
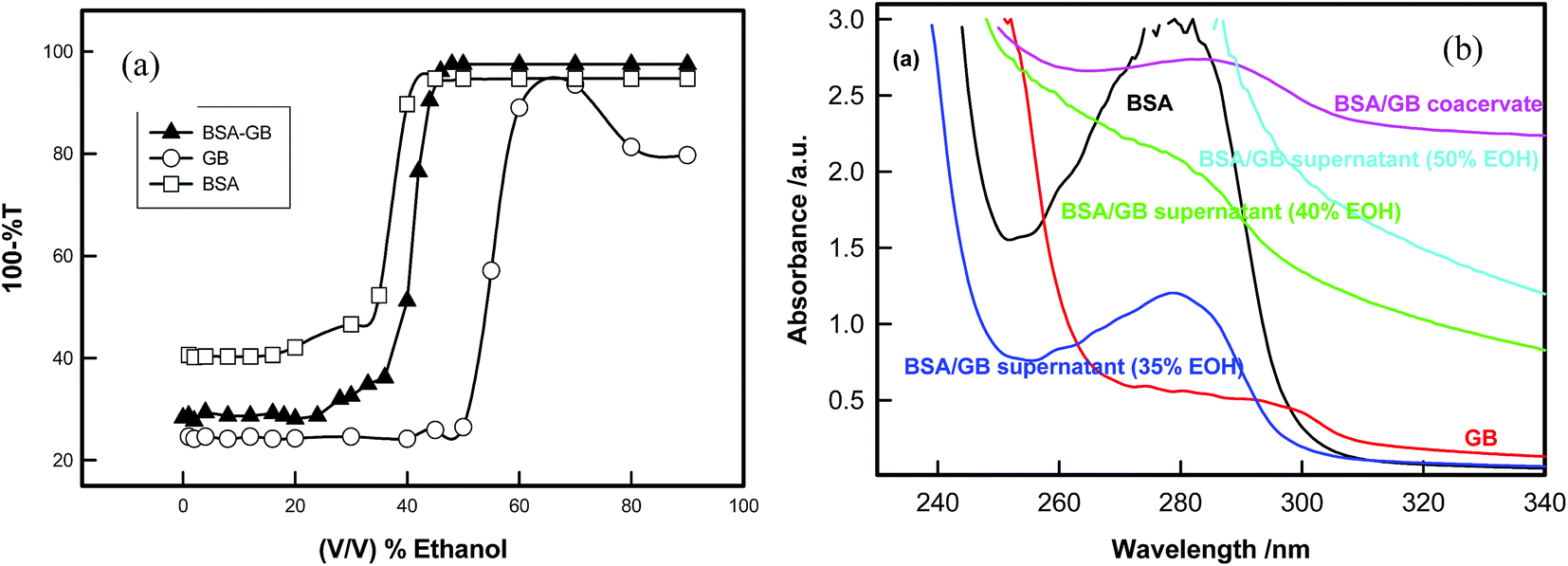 | ||
| Fig. 10 (a) Titration of BSA, GB and BSA/GB system in the presence of ethyl alcohol at pH (5.0 ± 0.1). (b) Far-UV-visible spectra of BSA, GB and BSA/GB in presence of different EOH in the wavelength range 240 to 340 nm. | ||
The SANS intensity profiles are plotted on a double logarithmic scale. In Fig. 8(b) it was found that all curve decrease sharply at different q ranges. It was found that in both curves intensity decreased sharply at small q range (0.001 < q < 0.05 Å−1), decreased marginally at intermediate q range (0.05 Å−1 < q < 0.12 Å−1), and eventually converged at large q range (q > 0.12 Å−1). The q-regions were separated manually by examining the I(q) versus q and I(q) versus q2 plots. A clear slope change was discernible at q = 0.05 Å−1 and 0.1 Å−1. The SANS data were analyzed in random phase approximation in different regimes. In low q-regime, data provided excellent least square fitting to power law.
| I(q) = IPL(0)q−α | (9) |
And at intermediate q region, the data could be fitted to the Ornstein–Zernike function57 given by
| I(q) = IOZ(0)/(1 + ξ2q2); ξq ⋘ 1 | (10) |
From the examination of SANS data the exponent, α defined by eqn (9) is known to owe its origin to the geometry of the scattering moiety in a given system. For instance, α = 1, 1.7, 2, and 4 correspond to geometrical shapes of rod, diffusion limited aggregation (DLA), Gaussian coil, and sphere, respectively. For self-similar objects, this exponent is equivalent to the mass fractal dimension of the object. In case of coacervates we found α = 1.07 ± 0.3, this value corresponds to the geometrical shape of rod in a given system at low-q regime and in the intermediate q-regime mesh size (ξ = 7.1 ± 0.2 nm) was determined from the experimental data using eqn (10). Thus, we concluded from SANS and turbidity data that the β-Lg had no influence on BSA/GB coacervation.
Fig. 9 shows the UV-visible and FT-IR spectra of BSA/β-Lg/GB supernatant and BSA, β-Lg and GB solutions which show that supernatant absorption profile are similar to β-Lg solution which indicated that the β-Lg was separated from BSA/β-Lg/GB mixture due to selective coacervation.
3.6 Removal of BSA
Finally for the removal of second protein (BSA) from the coacervate of BSA–GB system, we used ethyl alcohol for separating these two biopolymers. Both GB and BSA are not soluble in alcohols whereas water is a good solvent. As ethyl alcohol was added to this diluted coacervate solution water molecules got bound to alcohol molecule through hydrogen bonding and the resultant solvent mixture became a marginal solvent. The poor solvent quality compelled the GB molecule to reduce its spatial extension thereby bringing charge segment to each other's vicinity through electrostatic interaction. The GB molecules aggregated at the bottom (lower phase) and BSA remained in supernatant (upper phase).The titration of BSA/GB mixture in the presence of alcohol shows that phase separation occurs after the ethanol concentration ≈35% (v/v) and confirmation of presence of BSA in the supernatant was established by UV-visible and circular dichroism data (Fig. 10(a) and Fig. S4, ESI†). UV-vis spectra clearly indicated that supernatant absorbance spectra were exactly similar to native BSA absorbance spectra in 35% ethanol solution (Fig. 10(a)). In 35% EOH solution the BSA yield was 40%. Up to 50% EOH concentration, the secondary structure of BSA does not change very much (see Table 3) above that concentration, BSA gets denaturated.
| Samples | Helix/% | β-turn/% | Random coil/% | Antiparallel/% |
|---|---|---|---|---|
| BSA | 69.4 | 21.2 | 50.8 | 12.8 |
| 35% EOH | 54.4 | 21.6 | 51.4 | 12.6 |
| 40% EOH | 50.2 | 21.8 | 51.8 | 12.5 |
| 50% EOH | 40.1 | 22.1 | 52.4 | 12.2 |
After BSA–GB coacervation, β-Lg was present in the supernatant which was confirmed from UV-visible and FTIR spectra (see Fig. 9). Hence, β-Lg was separated in the first step. In the second step, for separating BSA from BSA–GB coacervate we used ethyl alcohol. After diluting BSA–GB coacervate and addition of ethyl alcohol, there was aqueous two-phase separation. The resulting mixture was centrifuged for 30 minute at 10000 rpm to produce dilute (upper) and dense (lower) phase. BSA was confirmed in the upper phase (supernatant) by UV-visible spectra (see Fig. 10).
4. Conclusion
The interaction with GB, a biocompatible polyampholyte, was used to isolate a target protein, bovine serum albumin (BSA), from mixture with a second protein of similar pI, β-lactoglobulin. Fig. 11 provides a schematic representation of the separation protocol. The crucial selection of optimum pH and mixing ratio for selective coacervation was guided by high sensitive turbidmitric titration, and to define condition for complex formation and coacervation. The interaction of GB with BSA and β-Lg was studied by fluorescence and circular dichroism spectroscopy. The binding of GB quenched the BSA and β-Lg fluorescence with corresponding binding constant. Fluorescence data indicated the binding constant of BSA/GB system was higher than β-Lg/GB and the distance between the acceptor GB, and the donor, BSA/β-Lg was estimated on the basis of Förster resonance energy transfer (FRET), this indicated that in BSA larger FRET efficiency (E) value was prevalent and the smaller r (distance between donor and acceptor) value indicated high energy transfer efficiency. In comparison, in β-Lg samples low E value and the larger r value indicated significantly low energy transfer efficiency. The fluorescence results were correlated with those obtained from circular dichroism data, which revealed change in protein conformation during the interaction process.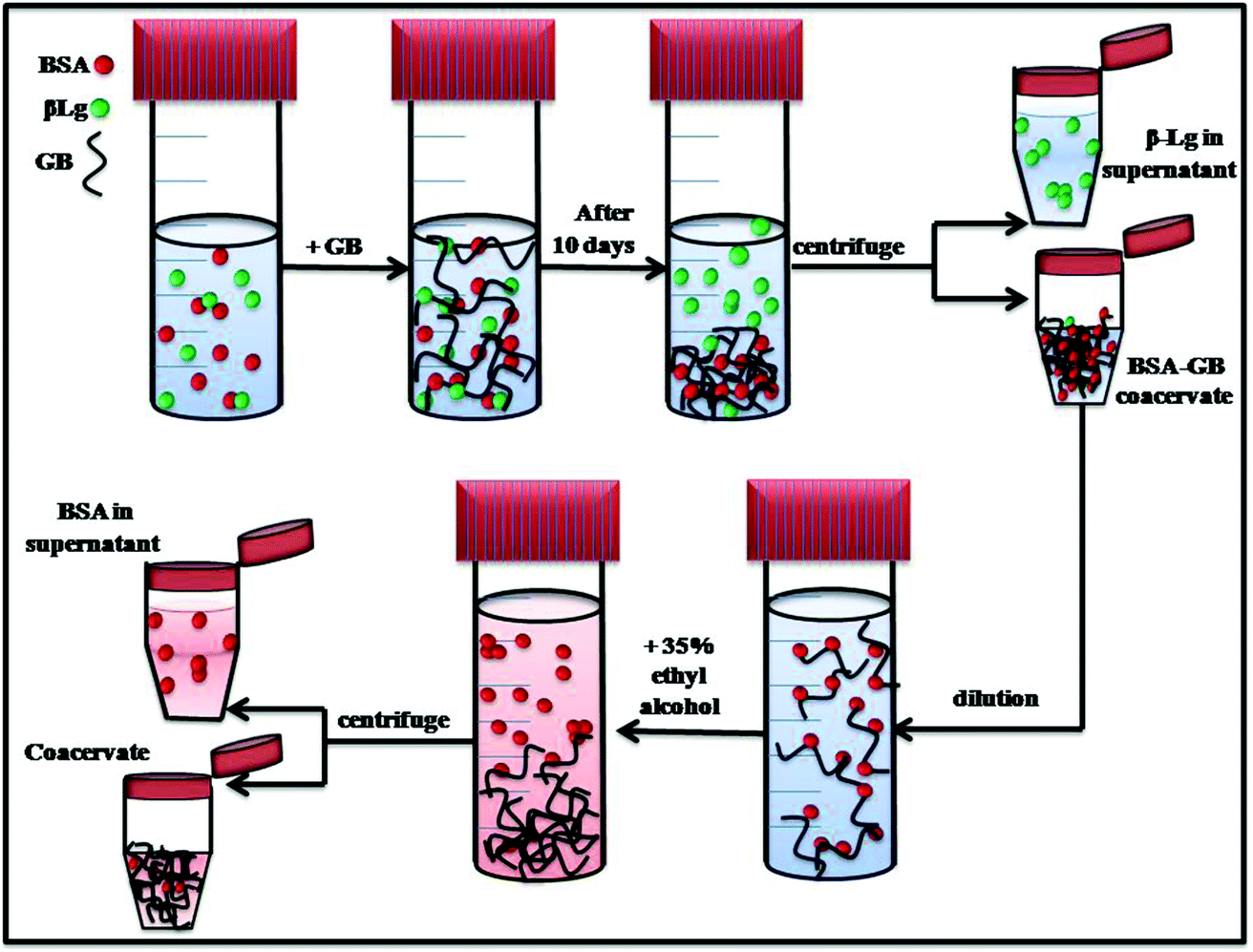 | ||
| Fig. 11 Representative model of protein–protein separation in a BSA–GB–β-Lg aqueous solution. | ||
Whereas the turbidmetric titration, fluorescence spectra and CD spectra show stronger binding of GB with BSA, the extent of separation possible is not evident from these results. From the titration of BSA–GB at different weight ratio and in the presence and absence of β-Lg, it may be seen that β-Lg had no influence on the BSA with GB. Also for the analysis of coacervate of BSA/GB in presence and absence of β-Lg by small angle neutron scattering shows, in both coacervate α = 1.07 ± 0.3, this value confirms to the geometrical shape of rod in a given system at low-q regime and at intermediate q-regime the mesh size ξ = 7.1 ± 0.2 nm. Thus, we conclude from SANS and turbidity data that the β-Lg had no influence on BSA/GB coacervate. The UV-visible and FT-IR spectra of BSA/β-Lg/GB supernatant and BSA, β-Lg and GB solutions which showed that the supernatant spectra was similar to β-Lg spectra. This indicated that only β-Lg was separated from the BSA/β-Lg/GB mixture after phase separation during coacervation. β-Lg was separated in first step while BSA remains in BSA–GB coacervate. For removal of BSA we used ethyl alcohol in diluted BSA–GB coacervate solution. The GB molecule tends to aggregated at the bottom and BSA remains in supernatant which is extracted by centrifugations. In summary, it is clearly demonstrated that complex coacervation is a suitable method for protein separation and purification.
Acknowledgements
JP acknowledges University Grants Commission, Government of India for a BSR-Research Fellowship. KR is thankful to Department of Science and Technology, Government of India-Inspire Faculty Award. This work was supported by a grant from Department of Science and Technology, Government of India. Authors are thankful AIRF of the University for providing access to CD spectrometer.References
- R. J. Stewart, C. S. Wang and H. Shao, Adv. Colloid Interface Sci., 2011, 167, 85–89 CrossRef CAS PubMed
.
- R. H. Tromp, C. G. de Kruif, M. V. Eijk and C. Rolin, Food Hydrocolloids, 2004, 18, 565–572 CrossRef CAS
- F. Sedlmeyer, M. Brack, B. Rademacher and U. Kulozik, Int. Dairy J., 2004, 14(4), 331–336 CrossRef CAS
- R. Tuinier, C. Rolin and C. G. de Kruif, Biomacromolecules, 2002, 3, 632–638 CrossRef CAS PubMed
- H. G. Bungenberg de Jong, Complex colloid systems, in Colloid Science, ed. H. R. Kruyt, Elsevier Publishing Company, Amsterdam, 1949, ch. X, vol. II, pp. 335–432 Search PubMed
- D. J. Clark and T. Kimura, J. Mol. Biol., 1990, 211, 883–896 CrossRef CAS PubMed
- J. Widom, Annu. Rev. Biophys. Biomol. Struct., 1998, 27, 285–327 CrossRef CAS PubMed
- J. Clausell, N. Rappel, T. K. Hale, D. Doenecke and M. Beato, PLoS One, 2009, 4, e0007243 Search PubMed
- N. Tavoosi, R. L. Davis-Harrison, T. V. Pogorelov, Y. Z. Ohkobo, M. J. Acario, M. C. Clay, C. M. Rienstra, E. Tajkhorshid and J. H. Morrissey, J. Biol. Chem., 2011, 286, 23247–23253 CrossRef CAS PubMed
- M. D. Golinska, T. T. H. Pham, M. W. T. Werten, F. A. de Wolf, M. A. Cohen Stuart and J. van der Gucht, Biomacromolecules, 2013, 14(1), 48–55 CrossRef CAS PubMed
- S. Boral and H. B. Bohidar, J. Phys. Chem. B, 2010, 114, 12027–12035 CrossRef CAS PubMed
- A. Gupta and H. B. Bohidar, J. Phys. Chem. B, 2007, 111, 10137–10145 CrossRef CAS PubMed
- C. Schmitt, C. Sanchez, D. Banon and S. Hardy, Crit. Rev. Food Sci. Nutr., 1998, 38, 689–753 CrossRef CAS PubMed
- J. L. Doublier, C. Garnier, D. Renard and C. Sanchez, Curr. Opin. Colloid Interface Sci., 2000, 5, 202–214 CrossRef CAS
- C. G. de Kruif, F. Weinbreck and R. de Vries, Curr. Opin. Colloid Interface Sci., 2004, 9, 340–349 CrossRef CAS
- T. V. Burova, N. V. Grinberg, I. A. Golubeva, A. Y. Mashkevich, V. Y. Grinberg and V. B. Tolstoguzov, Food Hydrocolloids, 1999, 13, 7–14 CrossRef CAS
- D. J. Burgess and J. E. Carless, J. Colloid Interface Sci., 1984, 98, 1 CrossRef CAS
- S. Wang, K. Chen, Y. Xu, X. Yu, W. Wang, L. Li and X. Guo, Soft Matter, 2013, 9, 11276–11287 RSC
- X. Du, P. L. Dubin, D. A. Hoagland and L. Sun, Biomacromolecules, 2014, 15, 726–734 CrossRef CAS PubMed
- X. Yisheng, M. Mazzawi, K. Chen, L. Sun and P. L. Dubin, Biomacromolecules, 2011, 12, 1512–1522 CrossRef PubMed
- Y. F. Wang, J. Y. Gao and P. L. Dubin, Biotechnol. Prog., 1996, 12, 356–362 CrossRef CAS
- Y. P. Fang, L. B. Li, C. Inoue, L. Lundin and I. Appelqvist, Langmuir, 2006, 22, 9532–9537 CrossRef CAS PubMed
- P. L. Dubin, J. Gao and K. W. Mattison, Sep. Purif. Methods, 1994, 23, 1–16 CrossRef CAS
- H. Yavuz and A. Denizli, Macromol. Biosci., 2004, 4, 84–91 CrossRef CAS PubMed
- R. Ghosh, J. Chromatogr., 2002, 952, 13–27 CrossRef CAS PubMed
- S. M. Smith, Protein Chromatography Methods in Molecular Biology, 2011, vol. 681, pp. 485–496 Search PubMed
- H. Morawetz and W. L. Hughes Jr, J. Phys. Chem., 1951, 56, 64 CrossRef
- R. R. Burgess and J. J. Jendrisak, Biochemistry, 1975, 14, 4636 Search PubMed
- M. A. Strege, P. L. Dubin, J. S. West, D. Flinta and M. Ladish, American Chemical Society, New York, 1990, ch. 5, vol. 427, pp. 66–79 Search PubMed
- M. Sternberg and D. Hershberger, Biochim. Biophys. Acta, 1974, 342, 195 CrossRef CAS
- J. Y. Shieh and C. E. Glatz, Polym. Prepr., 1991, 32, 606 CAS
- J. Xia and P. L. Dubin, Macromolecular Complexes in Chemistry and Biology, ed. P. L. Dubin, J. Bock, R. Davis, D. N. Schulz and C. Thies, Springer Verlag, Berlin, 1994, ch 5, p. 247 Search PubMed
- A. Toumadje, S. W. Alcorn and W. C. Johnson Jr, Anal. Biochem., 1992, 200, 321–331 CrossRef CAS PubMed
- V. K. Aswal and P. S. Goyal, Curr. Sci., 2000, 79, 947–953 Search PubMed
- Y. Wang, J. Y. Gao and P. L. Dubin, Biotechnol. Prog., 1996, 12, 356–362 CrossRef CAS
- P. L. Dubin, S. S. The, D. W. McQuigg, C. H. Chew and L. M. Gans, Langmuir, 1989, 5, 89–95 CrossRef CAS
- A. B. Kayitmazer, H. B. Bohidar, K. M. Mattison, A. Bose, J. Sarkar, A. Hashimoto, P. S. Russo, W. Jaeger and P. L. Dubin, Soft Matter, 2007, 3, 1064–1076 RSC
- K. Kaibara, T. Okazaki, H. B. Bohidar and P. L. Dubin, Biomacromolecules, 2000, 1, 100–107 CrossRef CAS PubMed
- K. Rawat, V. K. Aswal and H. B. Bohidar, J. Phys. Chem. B, 2012, 116, 14805–14816 CrossRef CAS PubMed
- K. Rawat and H. B. Bohidar, J. Phys. Chem. B, 2012, 116, 11065–11074 CrossRef CAS PubMed
- A. Tiwari, S. Bindal and H. B. Bohidar, Biomacromolecules, 2009, 10, 184–189 CrossRef CAS PubMed
- A. Gupta, B. Mohanty and H. B. Bohidar, Biomacromolecules, 2005, 6, 1623–1627 CrossRef CAS PubMed
- J. Pathak, K. Rawat and H. B. Bohidar, Int. J. Biol. Macromol., 2014, 63, 29–37 CrossRef CAS PubMed
- U. Kragh-Hansen, Pharmacol. Rev., 1981, 33, 17–53 Search PubMed
- H. H. Young, F. M. Herman, M. B. Norbert, G. O. Charles, M. Georg and I. Jacqueline, Inter science, 1967, 7, 446 CAS
- Principles of fluorescence spectroscopy, ed. J. R. Lakowicz, Kluwer Academic/Plenum publishers, Dordrecht, The Netherlands, 2004 Search PubMed
- W. R. Ware, J. Phys. Chem., 1962, 66, 455–458 CrossRef CAS
- Y. J. Hu, Y. Liu, A. X. Hou, R. M. Zhao, X. S. Qu and S. S. Qu, Acta Chim. Sin., 2004, 62, 1519–1523 CAS
- Y. J. Hu, Y. Liu, J. B. Wang, X. H. Xiao and S. S. Qua, J. Pharm. Biomed. Anal., 2004, 36, 915–919 CrossRef CAS PubMed
- A. R. Clapp, I. L. Medintz, J. M. Mauro, B. R. Fisher, M. G. Bawendi and H. Mattoussi, J. Am. Chem. Soc., 2004, 126, 301–310 CrossRef CAS PubMed
- L. A. Sklar, B. S. Hudson and R. D. Simoni, Biochemistry, 1977, 16, 819–828 CrossRef CAS PubMed
- L. Stryer, Annu. Rev. Biochem., 1978, 47, 819–846 CrossRef CAS PubMed
- J. N. Demas and G. A. Crosby, J. Phys. Chem., 1971, 75, 991–1024 CrossRef
- Z. C. Shang, P. G. Yi, Q. L. Yu and R. L. Lin, Acta Phys.-Chim. Sin., 2001, 17, 48–52 CAS
- X. X. Cheng, Y. Lui, B. Zhou, X. H. Xiao and Y. Liu, Spectrochim. Acta, Part A, 2009, 72, 922–1134 CrossRef PubMed
- L. Shang, Y. Wang, J. Jiang and S. Dong, Langmuir, 2007, 23, 2714–2721 CrossRef CAS PubMed
- P. G. de Gennes, Scaling Concepts in Polymer Physics, 2nd edn, Cornell University Press, Ithaca, NY, 1985 Search PubMed
- K. Okano, E. Wada, K. Kurita and H. Fukaro, J. Appl. Crystallogr., 1978, 11, 507–510 CrossRef CAS
- T. Cosgrove, S. J. White, A. Zarbakhsh, R. K. Heenan and A. M. Howe, Langmuir, 1995, 11, 744–749 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Phase boundaries of binding of BSA and β-Lg with GB, binding profile and coacervation yield of BSA–GB–β-Lg at various stoichiometric binding ratios. Far-UV CD-spectra of BSA and β-Lg in presence and absence of GB and ethanol undertaken in this study are included in the ESI. See DOI: 10.1039/c4ra13133a |
| This journal is © The Royal Society of Chemistry 2015 |