
A novel tin-based imidazolium-modified montmorillonite catalyst for the preparation of poly(butylene terephthalate)-based nanocomposites using in situ entropically-driven ring-opening polymerization†
Abstract
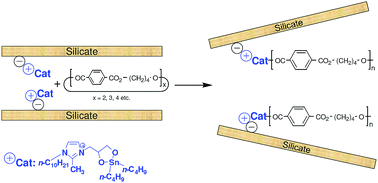
A novel alkylimidazolium salt incorporating a 2,2-di-n-butyl[1,3,2]dioxastannolane moiety was synthesized and characterized. Its effectiveness as a transesterification catalyst in the entropically-driven ring-opening polymerization (ED-ROP) of macrocyclic oligomers of butylene terephthalate (BT-MCOs) was comparable to that of other tin-based transesterification catalysts. A supported version of the catalyst was prepared by ion exchange with the sodium cations present in montmorillonite. X-Ray analysis indicated the imidazolium species entered the galleries between the silicate layers. The supported catalyst was significantly more thermally stable than the non-supported catalyst. Poly(butylene terephthalate)–clay nanocomposites were obtained by the in situ ED-ROP of BT-MCOs intercalated between the clay layers, catalyzed by either the supported tin-based catalyst or by a catalyst prepared in situ from a supported imidazolium salt with diol moieties and di-n-butyldimethoxytin. X-Ray diffraction and transmission electron microscopy indicated that the ensuing nanocomposites exhibit a mix of intercalated and exfoliated silicate nanolayers.
 Please wait while we load your content...
Please wait while we load your content...

