
Fluorescent diphenylfluorene-pyrenyl copolymer with dibenzothiophene-S,S-dioxide and adamantane units for explosive vapor detection†
Yuerong Wangab,
Yixun Gaoab,
Lei Chenab,
Yanyan Fua,
Defeng Zhua,
Qingguo He*a,
Huimin Caoa,
Jiangong Cheng*a,
Runsheng Zhangc,
Shuiqing Zhengc and
Songmao Yanc
aState Key Laboratory of Transducer Technology, Shanghai Institute of Microsystem and Information Technology, Chinese Academy of Sciences, 865 Changning Road, Shanghai 200050, China. E-mail: hqg@mail.sim.ac.cn; jgcheng@mail.sim.ac.cn; Fax: +86-21-62511070-8934; Tel: +86-21-62511070-8967
bUniversity of Chinese Academy of Sciences, 19 Yuquan Road, Beijing 100039, China
cShanghai Key Laboratory of Crime Scene Evidence, Shanghai Institute of Forensic Science, 803 North ZhongShan No. 1 Road, Shanghai 200083, China
First published on 5th December 2014
Abstract
A fluorescent diphenylfluorene-pyrenyl copolymer with dibenzothiophene-S,S-dioxide (SO) and adamantane units has been successfully synthesized via a Suzuki–Miyaura cross-coupling reaction. After studying the sensing properties of a series of diphenylfluorene-pyrenyl materials towards TNT vapor, it was found that the dibenzothiophene-S,S-dioxide (SO) units introduced into the diphenylfluorene-pyrenyl copolymers can simultaneously enhance the photostability and sensing performance of the fluorescent sensing materials. This simple strategy can be used as a promising approach for the development of fluorescent conjugated sensing materials.
1. Introduction
The detection of TNT and its analogues, which are widely used explosives causing serious security threats and health issues, has been studied for tens of years.1 The detection of TNT vapor is essential but more challenging because of its low volatility compared to that in solution and solid phases. Therefore, many research efforts have now been put into fluorescent conjugated polymers (FCPs) due to their higher sensitivity among the detection techniques and their “molecular wire” effect.2–4 However, FCPs films generally suffer from poor photostability, π–π stacking-induced aggregation in the solid state and low permeability to analytes, which restrict their sensing performance.5,6 Previously, some strategies have been prompted in molecular design and film morphology tuning to deal with the aggregation and permeability problems. For molecular structure modification, pentiptycene, hyper-branched, star-like and cross-linked architectures, bulky spacers and non-conjugated side groups have been introduced into polymer chains.6–10 For example, Nie et al. introduced a 4-[tris-(4-octyloxyphenyl)methyl]phenyl side chain to poly(2,7-carbazole) and demonstrated that the large and rigid side chains could reduce the interactions between polymer chains in the solid state, contributing to the sensitivity due to increased accessible space for the explosives.11 Similar findings were also reported by other groups.12 For film morphology tuning, spin-coating and dip-coating are most commonly used methods, and other approaches like electrospinning, self-assembled monolayers, molecular imprinting, and freeze drying were also employed to increase the area-to-volume ratio of sensing films, which could help to improve the sensing performance.13–17 Zhang et al. prepared a series of pyrene-functionalized films via monolayer assembly,which were revealed to be sensitive to trace amounts of nitroaromatic vapors.18 In addition, Xie et al. reported a surface molecular self-assembly strategy for molecular imprinting of polymer nanowires and nanotubes, which showed high capacity for binding TNT molecules.19 Recent studies on TNT vapor detection are mainly focused on making conjugated polymers into nanophases or porous phases to create more contact sites or pathways for TNT molecules. In those cases, to obtain high sensitivity and satisfactory selectivity is still challenging.20–23 Therefore, a simple strategy to enhance the sensing performance will be highly preferred.Dibenzothiophene-S,S-dioxide (SO) has recently been employed in organic photovoltaics (OPVs), organic light-emitting diodes (OLEDs) and organic field-effect transistors (OFETs) as an electron acceptor owing to its contribution to the spectral stability and fluorescence quantum efficiency improvement.24–26 The improved spectral stability was attributed to the electron-withdrawing –SO2– group in the SO unit in the backbone. The SO unit is also proven to be effective in lowering the LUMO energy level.27,28 Liu et al. incorporated a dibenzothiophene-S,S-dioxide (SO) isomer into a poly[9,9-bis(4-(2-ethylhexyloxy)phenyl)-fluorene-2,7-diyl] (PPF) backbone and investigated the electroluminescent properties.28 With an increasing number of SO units in the polymers, the LUMO levels of the copolymers decreased steadily. In addition, adamantane groups were introduced into the fluorescent conjugated polymer as side chains to effectively suppress interchain interactions and generate pathways or cavities for easier diffusion of explosive molecules into the sensing films.10
In this work, a conjugated diphenylfluorene-pyrenyl structure was chosen as the structural skeleton to guarantee that the LUMO level was higher than that of TNT. SO units were introduced for photostability improvement, affinity tuning and control of the interchain interactions of the conjugated polymers. Bulky caged adamantane groups were employed as pendent units to generate pathways or cavities for the effective diffusion of explosive molecules. Three fluorescent materials, including fluorene-pyrene copolymers without (P1) or with (P2) SO units, and their model compound M3, were designed and synthesized. The results of TNT vapor sensing indicated that the introduction of SO units benefits both the photostability and quenching efficiency of conjugated polymers to TNT vapor. The mechanism of these effects was systematically discussed.
2. Experimental section
2.1 Instruments
The 1H-NMR spectra were recorded on a Bruker DRX500 instrument, and tetramethylsilane (TMS) was used as an internal standard. The UV-Vis and fluorescence spectra were obtained using a Jasco UV- spectrophotometer and a Jasco FP 6500 spectrometer, respectively. The quantum yields of the film were measured using a Jasco FP 6600 spectrofluorometer with an integrating sphere and excitation wavelengths for P1 and P2 of 391 and 373 nm, respectively. Cyclic voltammetry (CV) experiments were performed usingh a CH Instruments electrochemical analyzer. The electrochemical properties of the materials were investigated by CV with a standard three-electrode (a glassy carbon electrode as a working electrode, a platinum electrode as a counter electrode and a saturated calomel electrode as a reference electrode) electrochemical cell in a 0.1 M Bu4NPF6 in acetonitrile solution under nitrogen atmosphere with a scanning rate of 100 mV s−1 at room temperature. Pristine P1, P2 and M3 films on (10 × 20 mm) quartz plates were spin-coated at 2000 rpm from their solutions and placed under vacuum for 30 minutes before use. The fluorescent responses of P1, P2 and M3 films to TNT vapor and other explosives were performed at room temperature in hermetically-sealed cuvettes containing cotton and analytes, with the cotton on the analytes to prevent direct contact between the films and analytes and to help maintain a stable vapor pressure. The fluorescence time-course responses were recorded as soon as the quartz plate was exposed to the analyte vapor.2.2 Synthesis
The synthetic procedures are illustrated in Schemes 1 and 2. The fluorescent materials P1, P2 and M3 were synthesized via Suzuki–Miyaura cross-coupling reactions with yields of 60%, 52% and 86%, respectively. The starting materials, including adamantane substituted (2,7-dibromo-9,9-bis(4-octyloxy-phenyl))fluorene (3), 2,7-bis(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-1,6-dihydropyrene and 3,7-dibromo-dibenzothiophene-S,S-dioxide (SO), were prepared according to the literature.10,24 Commercially available reagents were used as received without further purification.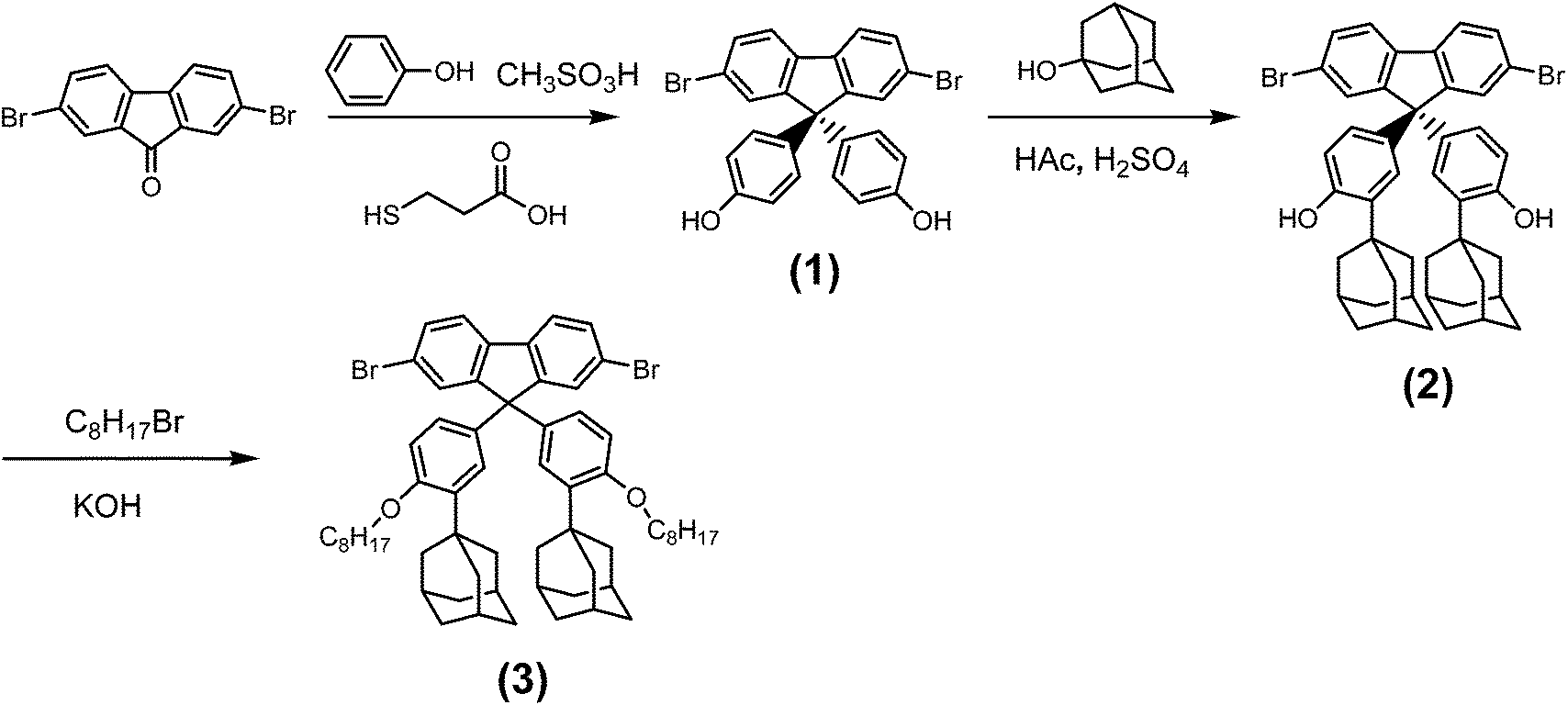 | ||
| Scheme 1 The synthesis path of fluorene 3. | ||
 | ||
| Scheme 2 The synthesis path of P1, P2 and M3. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 015, PDI = 2.24.
015, PDI = 2.24.3. Results and discussion
3.1 Optical properties
The normalized absorption and emission spectra of P1, P2 and M3, in THF solution and in films, are shown in Fig. 1. In THF solution, the absorption peaks of P1, P2 and M3 are at 346, 346 and 365 nm, and the fluorescence emission peaks are at 426, 426 and 421 nm, respectively. The absorption peaks of the P1, P2 and M3 films are at 391, 354 and 369 nm, and the fluorescence emission peaks are at 437, 441 and 433 nm, respectively. In THF solution, P1, P2 and M3 are highly fluorescent with quantum yields of 0.49, 0.40 and 1 (Table 1), respectively. | ||
| Fig. 1 The UV-Vis absorption and emission spectra of P1, P2 and M3 in solution (THF: 10−3 M, left) and in films (toluene: 10−3 M, right). | ||
| Solution (THF) | Film | HOMO (eV) | LUMO (eV) | ΔE (eV) | Φ | |||
|---|---|---|---|---|---|---|---|---|
| Abs λmax (nm) | PL λmax (nm) | Abs λmax (nm) | PL λmax (nm) | |||||
| a Against 9,10-diphenylanthracene in THF solution (Φ = 1, λem = 390 nm). | ||||||||
| P1 | 346 | 426 | 391 | 437 | −5.77 | −3.77 | 2.00 | 0.49 |
| P2 | 346 | 426 | 354 | 441 | −5.73 | −3.83 | 1.90 | 0.40 |
| M3 | 365 | 421 | 369 | 433 | −5.46 | −3.47 | 1.99 | 1 |
As is shown in Fig. 1, among the three materials, M3 shows the longest absorption peak in THF solution. This could be related to the pyrenyl unit, since its ratio to fluorene is 2:1 for M3, 1:1 for P1 and 1:0.7 for P2.
It could also be seen that, after the incorporation of SO units, the absorption and emission peaks of P2 in THF solution show little difference from those of P1. From solution to film, the absorption and emission peaks of P2 red-shifted by 8 and 15 nm, respectively. However, the absorption and emission peaks of P1 red-shifted by 45 and 11 nm from solution to film, respectively. Furthermore, the P1 film shows a special broad tail peak in its absorption spectrum, expanding from 400 to 600 nm, which cannot be found in the P2 film. The absorption spectral difference between P1 and P2 could be attributed to the fact that there exist strong interchain π–π stacking interactions in the P1 film but not in P2 film, which is commonly found in pyrenyl-containing materials because of its large aromatic ring and good excimer-forming capacity. The strong π–π stacking interaction, on one hand, usually results in fluorescence self-quenching, and on the other hand, will provide less contact sites for analytes and hence lead to a decrease in sensing performance.29 For further comparison, the quantum yields of P1 and P2 in film were also measured, using a spectrofluorometer with integrating sphere, and found to be 2.24% and 3.14%. It can be seen that P2 has a larger quantum yield in the solid state than P1. Thus, the incorporation of SO units helps to prevent the strong π–π stacking interaction in the P2 film.
3.2 Electrochemical properties
According to the photoinduced electron transfer (PET) mechanism, to detect TNT vapor, the LUMO level of the sensing probe should be higher than that of TNT in order to cause the fluorescence quenching process after excitation (Fig. 2). The oxidation and reduction potentials of both probes and analytes were obtained via cyclic voltammetry (CV) (Fig. S1†). The HOMO, LUMO and band gap data were calculated according to the oxidation and reduction potentials obtained and are summarized in Table 1. | ||
| Fig. 2 HOMOs and LUMOs of P1, P2 and M3, and their PET process to TNT. | ||
Since the LUMO levels of P1 (−3.77 ± 0.020 eV), P2 (−3.83 ± 0.026 eV) and M3 (−3.47 ± 0.031 eV) are all higher than that of TNT (−4.04 eV), all three materials could be theoretically used for TNT sensing based on the PET mechanism (Fig. 2). Compared with M3, P1 and P2 have lower LUMO levels because of the electron delocalization along the conjugated chains. Moreover, P2 has a relatively lower LUMO level than P1 owing to the presence of the electron-withdrawing SO units in its backbone.
Therefore, the introduction of SO units could not only decrease the strong π–π stacking interaction between polymer chains in the solid state, but could also finely tune the energy level.
3.3 Sensing properties
The relationship between film configuration (for example, by varying the concentration of the solution used for spin-coating, solvents and film fabrication methods) and sensing properties was studied. The results indicated that the solvents and film fabrication methods have little effect on the sensing performance of P1 and P2 (Fig. S2†), while they would greatly affect that of M3. Although the π–π stacking interactions in the P1 film are stronger than those of P2 as indicated from their absorption spectra, it is insufficient for formation of agglomerates in the P1 film like those observed in the M3 film using scanning electron microscopy (SEM) (Fig. 3). Films fabricated from different concentrations of solutions were investigated to optimize the sensing conditions for P1 and P2 films (Fig. 4). In all cases, the P2 film demonstrates a better sensing performance than the P1 film upon exposure to saturated TNT vapor, and for both P1 and P2, the best concentration of solution for spin-coating is 10−3 M in toluene. | ||
| Fig. 3 The surface topography images of P1 and P2 films (spin-coated; toluene; 10−3 M; 1.0 μm) and M3 films (spin-coated; 10−3 M; (a) toluene; (b) tetrahydrofuran; (c) dichloromethane) obtained using SEM. All the scale bars are 500 nm. | ||
 | ||
| Fig. 4 Quenching efficiencies of P1, P2 and M3 films in different concentrations (spin-coated; toluene: 10−5 M, 10−4 M, 10−3 M, 2 × 10−3 M) exposed to saturated TNT vapor for 300 s. | ||
Fig. 5 shows the sensing properties of P1, P2 and M3 films exposed to saturated TNT vapor (5 ppb) and air.30 In neat air, the P2 and M3 films exhibited excellent photostabilities under the excitation of their absorption maxima, but P1 was relatively unstable (an additional 14% was bleached compared to the P2 film after 300 s exposure). This could be attributed to the electron-withdrawing effect of the SO units of the P2 film, which reduce the electron density of the backbone and make it more able to resist photooxidation.28
 | ||
| Fig. 5 Sensing properties of P1, P2 and M3 films (spin-coated; toluene: 10−3 M) exposed to saturated TNT vapor and air for 300 s. | ||
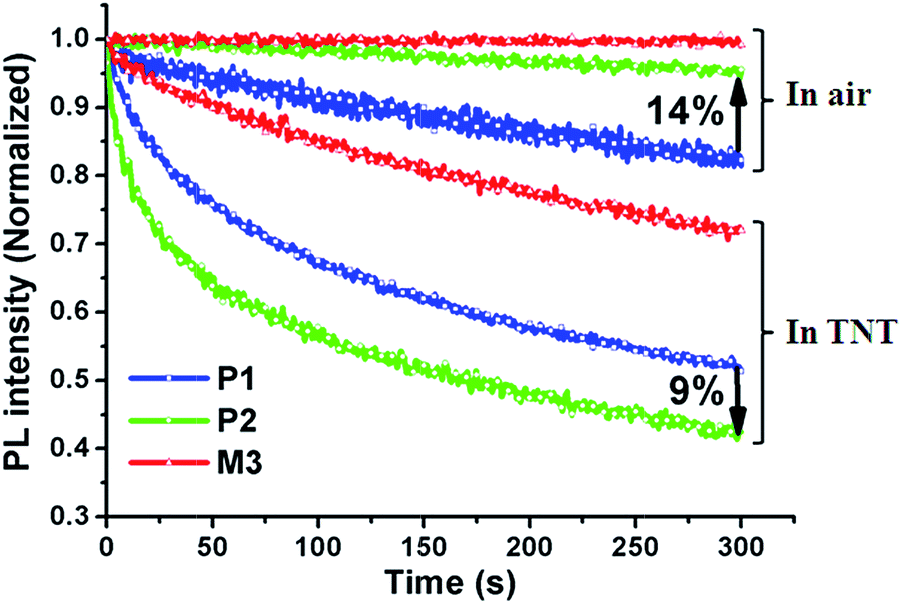
The emission intensities of the P1, P2 and M3 films were quenched by 24%, 36% and 10%, respectively, after 50 s, and by 49%, 58%, and 28%, respectively, after 300 s upon exposure to saturated TNT vapor. Obviously, the P2 film shows the highest quenching efficiency. The better sensing performance of the P2 film compared to the P1 film can be attributed to the incorporation of the electron-withdrawing SO units for two reasons. One reason is that the polar SO units could improve the polarity of the polymer, thus increasing the affinity between the polymer and the polar TNT molecules. The other is that the SO units could reduce the strong π–π stacking interactions between the polymer chains, thus providing more contact sites for analytes. Compared with P1 and P2, the small molecule M3 shows the poorest quenching efficiency, which in one aspect results from its lack of “molecular wire” effect that usually exists in conjugated polymers leading to an amplified fluorescence signal, and in another from the serious aggregation of the M3 film (Fig. 3(a)) which gives rise to fewer contact sites for TNT molecules. In addition, the fluorescence emission peak positions of the P1, P2 and M3 films show no change after exposure to TNT vapor, indicating a PET mechanism between TNT and the sensing materials. For the M3 film, it was also found that solvents (tetrahydrofuran, toluene, dichloromethane) seriously influenced the sensing properties (Fig. 6). The highest quenching efficiency (40%) for the M3 film in TNT vapor was achieved from the tetrahydrofuran solution, while the lowest was obtained from its dichloromethane solution. As is shown in the SEM images (Fig. 3), the influence of the morphology on the quenching efficiency is caused by differing degrees of aggregation. The more serious the aggregation, the worse the quenching efficiency is. In addition, the optimized sensing property of the M3 film was still worse than that of P1 and P2.
 | ||
| Fig. 6 Quenching efficiencies of M3 films (blue: spin-coated; yellow: dip-coated; solution (from left to right): toluene, tetrahydrofuran, dichloromethane; concentration: 2 × 10−3 M, 10−3 M, 10−4 M) exposed to saturated TNT vapor for 300 s. | ||
As discussed above, the affinity between the sensory materials and TNT, the contact sites in the materials for TNT vapor, and the “molecular wire” effect in the polymers will together influence the sensing performance.
The reversibility of the quenching of the P2 film was determined by the sequential exposure of the film to saturated TNT vapor (5 ppb) and air for 60 s (Fig. 7). It was found that the fluorescence of the sensing film could be rapidly recovered, and after several cycles there is only a little attenuation, suggesting a good reversibility of the P2 film.
 | ||
| Fig. 7 Cycles of photoluminescence (PL) quenching and recovery by exposing the sensing film to saturated TNT vapor and air for 60 s sequentially. | ||
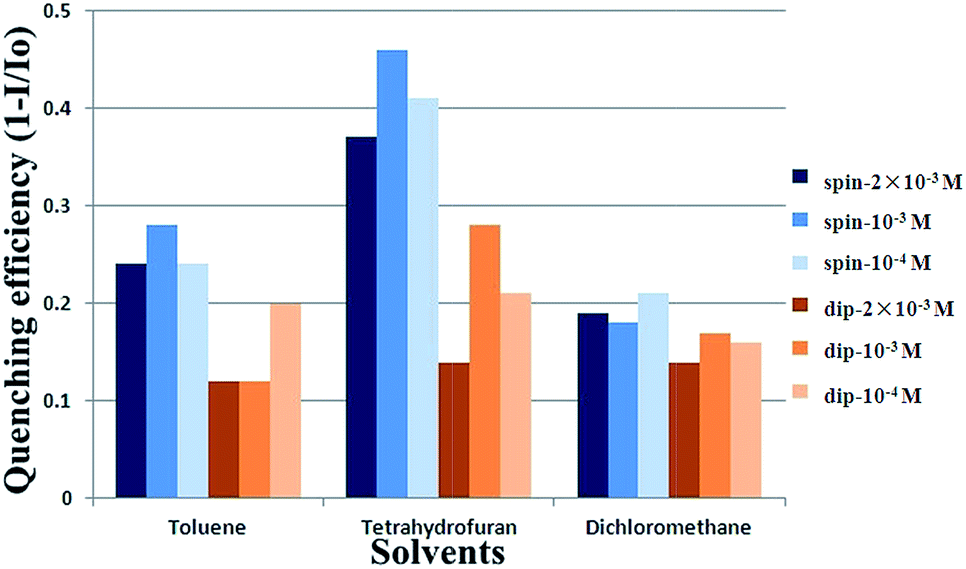
Fig. 8 presents the selectivity of the P2 film to several kinds of explosives. The P2 film shows excellent sensitivity to the saturated vapor of nitroaromatic explosives (NACs) including 4-NT (4-nitrotoluene), DNT (2,4-dinitrotoluene) and TNT with quenching efficiencies of 86%, 73%, and 58% within 300 s, respectively. However, it is only slightly quenched with some other explosives like 4-NP (4-nitrophenol), RDX, TATP (triacetone triperoxide) and PETN (pentaerythritol tetranitrate). This indicates that the PET process does not occur between P2 and other explosives that have higher LUMO levels than P2, like RDX (Fig. 9). In contrast, the LUMO levels of NACs (TNT, −4.04 eV; DNT, −3.84 eV; 4-NT, −4.16 eV, calculated from the CV curves shown in Fig. S1†) are all lower than that of P2, which guarantees the occurrence of the PET process.31 Furthermore, it was found that the quenching efficiencies with NACs increase with an increase in the saturated vapor pressure of NACs from 5 ppb to 215 ppm (TNT, 5 ppb; DNT, 290 ppb; 4-NT, 215 ppm) at room temperature.30 The results indicate that the P2 film shows satisfactory selectivity toward NACs. Furthermore, the effect of vapors of common solvents on the sensing performance of the P2 film was also checked (Fig. S3†). It was shown that the P2 film is quite stable or only slightly quenched in the saturated vapor of common solvents including toluene, tetrahydrofuran, dichloromethane, acetic ether, acetone and ethanol.
 | ||
| Fig. 8 Quenching efficiencies of P2 film (spin-coated; toluene: 10−3 M) exposed to the saturated vapor of common explosives for 300 s. | ||
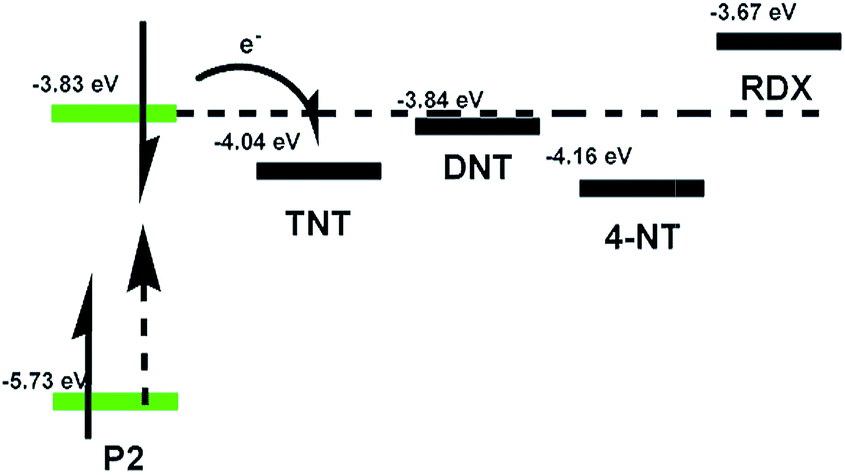 | ||
| Fig. 9 LUMOs of TNT, DNT, 4-NT and RDX and the PET process of P2 to them. | ||
4. Conclusion
In summary, a simple and effective approach was proposed towards nitroaromatic sensing materials by incorporating dibenzothiophene-S,S-dioxide (SO) units and adamantane units into the diphenylfluorene-pyrenyl backbones. After the incorporation of SO units, the stronger affinity of the sensory materials towards TNT and the effective prevention of aggregation resulting in more contacting sites for TNT vapor in the polymer films will together help to improve the sensing performance of the conjugated polymers. The SO units could also be useful in elevating the photostability of the polymers by reducing the electron density in the polymer backbones, allowing them to resist photooxidation.Compared with reported results, the simple strategy we used can bring about relatively good sensing performance and satisfactory selectivity of sensory materials for NACs. Further work is still required to achieve a higher quenching efficiency and rapid response to TNT vapor. To obtain much higher sensitivity, one method is further optimization of the ratio of SO to P2, which may increase the interaction force between the sensory material and TNT molecules and hence increase the sensitivity. Another way is to prepare a nano and porous film, which will increase the area to volume ratio of the sensory film and hence improve the sensitivity; this will be our next goal. This proposed strategy can be used as a promising approach to the development of fluorescent conjugated sensing materials.
Acknowledgements
The authors thank the research programs from the National Natural Science Foundation of China (no. 21273267, 61325001, 61321492 and 51473182), Ministry of Science and Technology of China (program no. 2012BAK07B03), and Shanghai Science and Technology Committee (grant no. 11JC1414700).Notes and references
- M. Krausa and A. A. Reznev, Vapour and Trace Detection of Explosives for Anti-terrorism Purposes, Kluwer Academic Publishers, Boston, 2004 Search PubMed.
- T. M. Swager, Acc. Chem. Res., 1998, 31, 201–207 CrossRef CAS.
- J. W. Gardner and J. Yinon, Electronic Nose & Sensors for the Detection of Explosives, Kluwer, Academic Publishers, 2003 Search PubMed.
- C. Deng, Q. He, C. He, L. Shi, J. Cheng and T. Long, J. Phys. Chem. B, 2010, 114, 4725–4730 CrossRef CAS PubMed.
- K. Brunner and J. W. Hofstract, Macromolecules, 2003, 36, 500–507 CrossRef.
- J. S. Yang and T. M Swager, J. Am. Chem. Soc., 1998, 120, 11864–11873 CrossRef CAS.
- J. Du, Q. Fang, D. Bu, S. Ren, A. Cao and X. Chen, Macromol. Rapid Commun., 2005, 26, 1651–1656 CrossRef CAS.
- W. J. Lin, W. C. Chen, W. C. Wu, Y. H. Niu and A. K. Y. Jen, Macromolecules, 2004, 37, 2335–2341 CrossRef CAS.
- Q. Zhao and W. Wu, Polymer, 2009, 50, 998–1004 CrossRef CAS PubMed.
- H. F. Leng and W. H. Wu, React. Funct. Polym., 2012, 72, 206–211 CrossRef CAS PubMed.
- H. Nie, Y. Zhao, M. Zhang, Y. Ma, M. Baumqarten and K. Müllen, Chem. Commun., 2011, 47, 1234–1236 RSC.
- J. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 21, 11864–11873 CrossRef.
- A. Greiner and J. H. Wendorff, Angew. Chem., Int. Ed., 2007, 46, 5670–5703 CrossRef CAS PubMed.
- K. Y. Hua, C. M. Deng, C. He, L. Q. Shi, D. F. Zhu, Q. G. He and J. G. Chen, Chin. Chem. Lett., 2013, 24, 643–646 CrossRef CAS PubMed.
- M. J. Shin, M. Yang and J. S. Shin, Part. Sci. Technol., 2012, 30, 543–552 CrossRef CAS.
- D. Nie, D. Jiang, D. Zhang, Y. Liang, Y. Xue, T. Zhou, L. Jin and G. Shi, Sens. Actuators, B, 2011, 156, 43–49 CrossRef CAS PubMed.
- W. E. Lee, C. J. Oh, I. K. Kang and G. Kwak, Macromol. Chem. Phys., 2010, 211, 1900–1908 CrossRef CAS.
- S. Zhang, F. Lu, L. Gao, L. Ding and Y. Fang, Langmuir, 2007, 23, 1584–1590 CrossRef CAS PubMed.
- C. Xie, Z. Zhang, D. Wang, G. Guan, D. Gao and J. Liu, Anal. Chem., 2006, 78, 8339–8346 CrossRef CAS PubMed.
- J. L. Novotney and W. R. Dichtel, ACS Macro Lett., 2013, 2, 423–426 CrossRef CAS.
- H. Nie, Y. Lv, L. Yao, Y. Pan, Y. Zhao, P. Li, G. Sun, Y. Ma and M. Zhang, J. Hazard. Mater., 2014, 264, 474–480 CrossRef CAS PubMed.
- Y. Wang, A. La, Y. Ding, Y. Liu and Y. Lei, Adv. Funct. Mater., 2012, 22, 3547–3555 CrossRef CAS.
- Y. Long, H. Chen, H. Wang, Z. Peng, Y. Yang, G. Zhang, N. Li, F. Liu and J. Pei, Anal. Chim. Acta, 2012, 744, 82–91 CrossRef CAS PubMed.
- C. Li, M. Liu, N. G. Pschirer, M. Baumgarten and K. Müllen, Chem. Rev., 2010, 110, 6817–6855 CrossRef CAS PubMed.
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed.
- S. M. Goetz, C. M. Erlen, H. Grothe, B. Wolf, P. Lugli and G. Scarpa, Org. Electron., 2009, 10, 573–580 CrossRef CAS PubMed.
- Y. Li, H. Wu, J. Zou, L. Ying, W. Yang and Y. Cao, Org. Electron., 2009, 10, 901–909 CrossRef CAS PubMed.
- J. Liu, J. Zou, W. Yang, H. Wu, C. Li, B. Zhang, J. Peng and Y. Cao, Chem. Mater., 2008, 20, 4499–4506 CrossRef CAS.
- C. He, Q. He, C. Deng, L. Shi, Y. Fu, H. Cao and J. Cheng, Synth. Met., 2011, 161, 293–297 CrossRef CAS PubMed.
- K. J. Albert and D. R. Walt, Anal. Chem., 2000, 72, 1947–1955 CrossRef CAS.
- H. Sohn, R. M. Calhoun, M. J. Sailor and W. C. Trogler, Angew. Chem., Int. Ed., 2001, 40, 2104–2105 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: CV curves and quenching efficiencies of P1 and P2 films prepared using different solvents. See DOI: 10.1039/c4ra12966k |
| This journal is © The Royal Society of Chemistry 2015 |