
Hierarchical self-assembly, photo-responsive phase behavior and variable tensile property of azobenzene-containing ABA triblock copolymers†
Abstract
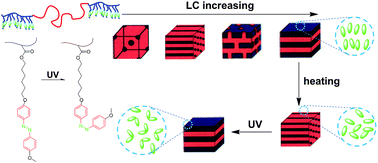
A series of triblock copolymers with liquid crystalline (LC) poly{6-[4-(4-methoxyphenylazo)phenoxy]hexyl methacrylate} (PMMAZO) as the end blocks and rubbery poly(n-butyl acrylate) (PnBA) as the midblock were synthesized. The effect of the interplay between the LC ordering and microphase separation on the hierarchical assembly of the triblock copolymers was studied. It is found that microphase separation at a larger scale can affect the LC ordering at a smaller scale, such as the stacking of the LC moieties, LC temperature and the domain size of the LC phase. On the other hand, alteration of the LC ordering, such as isotropization and smetic-to-nematic transition, may also lead to an order–order transition (OOT) or change in the long period of the microphase-separated structure. UV light can trigger the isomerization of the azobenzene LC moieties, which can be further amplified and exerts an effect on the microphase separation behavior, including the regularity of the microphase-separated structure and the OOT. The triblock copolymers also exhibit light-variable tensile properties. The results reveal that the phase behavior and mechanical properties of this type of triblock copolymer can be readily regulated by light, thus it may be used as smart and functional thermoplastic elastomer.
 Please wait while we load your content...
Please wait while we load your content...

