
2,5-Dithienylpyrrole (DTP) as a donor component in DTP–π–A organic sensitizers: photophysical and photovoltaic properties†
Walid Sharmoukh‡
ab,
Antonio Attanzioc,
Eva Busattoc,
Thibaud Etienneabd,
Stefano Carlic,
Antonio Monariab,
Xavier Assfeldab,
Marc Beleyab,
Stefano Caramori*c and
Philippe C. Gros*ab
aPhotosens, Université de Lorraine, UMR SRSMC, Vandoeuvre-Lès-Nancy, Nancy, 54506, France
bPhotosens, CNRS, UMR SRSMC, Vandoeuvre-Lès-Nancy, 54506, France. E-mail: philippe.gros@univ-lorraine.fr
cDepartment of Chemical and Pharmaceutical Sciences, University of Ferrara, Ferrara, 44121, Italy. E-mail: cte@unife.it
dUCPTS, Université de Namur, Rue de Bruxelles 61, 5000 Namur, Belgium
First published on 5th December 2014
Abstract
Organic dyes have been prepared to evaluate the ability of 2,5-dithienylpyrrole (DTP) to act as a donor substituent in D–π–A sensitizers for DSSCs. Using a styryl π-bridge the dyes were found to be excellent sunlight harvesters when adsorbed on TiO2 photoanodes with absorbances >3 in the 300–550 nm region. Calculations as well as transient absorption spectroscopy in both solution and on a TiO2 surface revealed that they were favourable for efficient injection and regeneration.
Introduction
The development of renewable energy is a current challenge to decrease the demand for fossil resources. In this context the harvesting of the abundant solar energy and its conversion into electricity is actively developed. Dye-sensitized solar cells (DSSCs)1 are promising photovoltaic devices because of their low cost and easily tunable color making them highly appropriate for zero-energy buildings. The principle of these DSSCs is to collect photons from sunlight using a sensitizer or dye, linked via electron-withdrawing groups to an anode (coated with nanocrystalline TiO2 semiconductor). The absorbed photons induce excitation of the dye and fast electron injection into the TiO2. A redox electrolyte or mediator then regenerates the oxidized dye in its ground state. The oxidized form of the mediator is then regenerated by reduction at the cathode, thus creating an electric circuit. The role of the dye is pivotal in both harvesting and transfer processes. DSSCs including Ru-dyes (N719 or black dye), gave impressive efficiency values in the 9–11% range.2a However ruthenium is a relatively scarce metal at the earth surface and could rapidly prevent large scale development and applications. Consequently metal-free dyes based on D–π–A systems with efficient light-driven intramolecular charge transfer has attracted the attention. Many kinds of organic dyes have been produced and have been compiled recently.3 The most common organic dyes contain an electron donor (D), usually triphenylamine known to stabilize holes generated upon irradiation, a π-delocalized bridge to ensure light harvesting and charge transport and an electron acceptor (A) for charge transfer and grafting to TiO2 surface (usually a cyanoacrylic acid).4 Despite better oxidation capability and enhanced luminescence properties,5 2,5-dithienylpyrrole (DTP) have been less studied6 for organic electronic applications than oligothiophenes.7 Recently, we have reported that bipyridine ligands bearing DTP as chromophoric substituents promoted interesting optical and electronic properties such as high molar extinction coefficients and wide absorption ranges to the corresponding homoleptic ruthenium complexes.8 Recently, Ho Hyun and coworkers have reported the use of 2,5-DTP as π-conjugated system in D–π–A dyes associated with triphenylamine as the donor (D).9 Taking into account its oxidation capability, DTP can be considered as a potential donor substituent (D), its conjugated structure also allowing an additional beneficial effect on absorption properties. Herein we report the preparation, characterization and photovoltaic performance of DTP–π–A dyes where a styryl π-spacer has been attached to the pyrrole (DTP1) or to the thiophene (DTP2) on the DTP moiety (Scheme 1).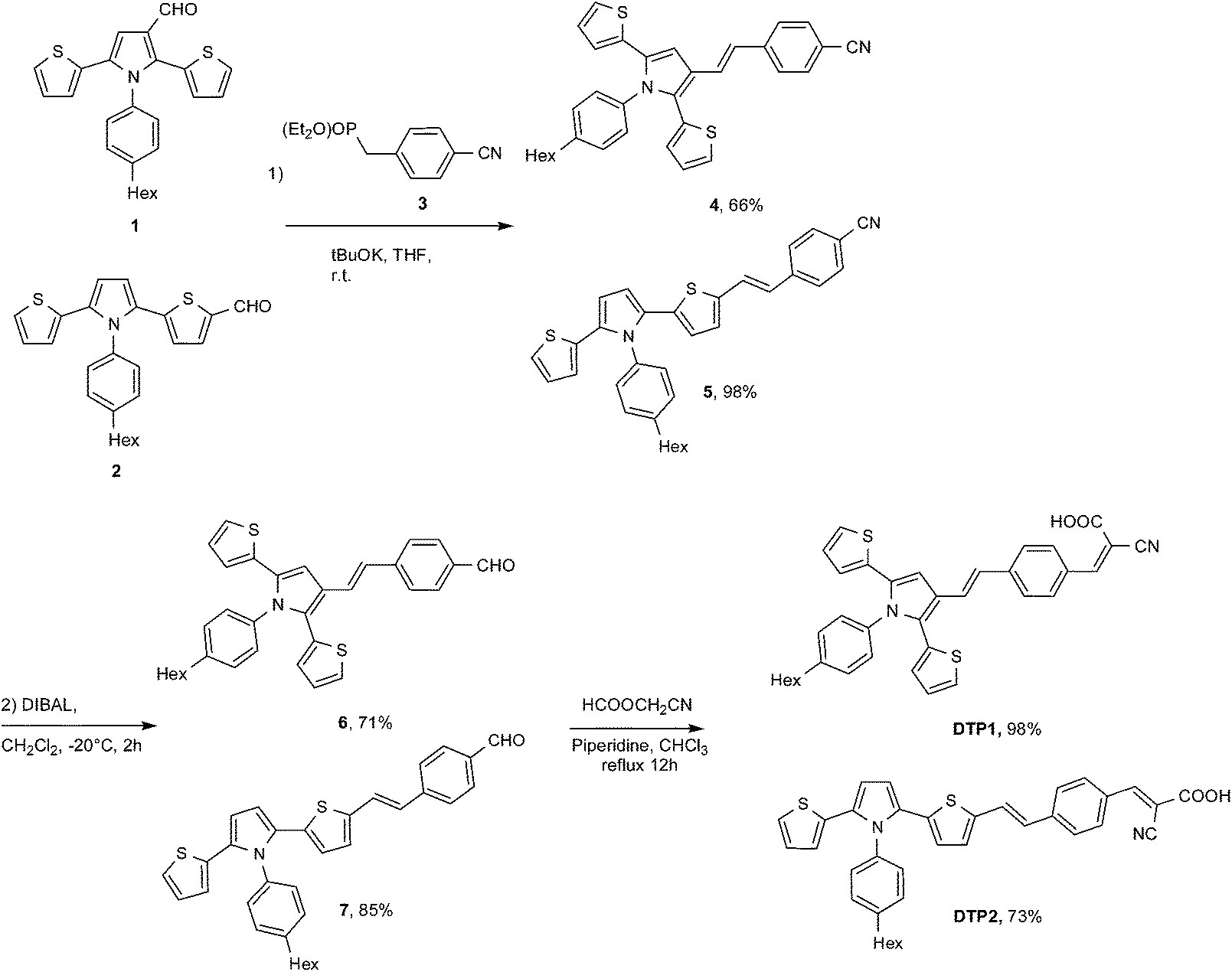 | ||
| Scheme 1 Synthesis of dyes DTP1 and DTP2. | ||
Results and discussion
Synthesis
The dyes DTP1 and DTP2 were synthesized using the Wadsworth–Emmons reaction as key reaction. This route required diethylphosphonate 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 and aldehydes 1 and 2 bearing the carbonyl function on pyrrole and thiophene respectively.8 Aldehydes 6 and 7 were respectively obtained by reaction of 1 and 2 with 3 in the presence of tBuOK as a base followed by the reduction of nitriles 4 and 5 with di-isobutyl aluminum hydride (DIBAL) in dichloromethane.
10 and aldehydes 1 and 2 bearing the carbonyl function on pyrrole and thiophene respectively.8 Aldehydes 6 and 7 were respectively obtained by reaction of 1 and 2 with 3 in the presence of tBuOK as a base followed by the reduction of nitriles 4 and 5 with di-isobutyl aluminum hydride (DIBAL) in dichloromethane.
The target dyes were DTP1 and DTP2 were finally obtained in respectively 98 and 73% yield by reacting the 6 and 7 with cyanoacetic acid using piperidine as a base (Scheme 1).
Spectroscopic and electrochemical characterization
The electronic properties of the new dyes were investigated by UV-vis absorption (Fig. 1) and fluorescence emission spectroscopy as well as cyclic voltammetry (Table 1 for data). | ||
| Fig. 1 UV-vis spectra of dyes in dichloromethane. | ||
| Dye | Absorption | Emission | Electrochemical data | ||||
|---|---|---|---|---|---|---|---|
| λmaxa (nm) | ε (M−1 cm−1) | λmaxb (nm) | Eoxc (V) | Eredc (V) | E0–0d (eV) | E*oxe (V) | |
| a Measured in CH2Cl2 at 25 °C.b Photomultiplier corrected emission maxima for the complexes in CH2Cl2 A < 0.05 in the absence of O2.c Recorded in CH2Cl2 using Bu4N+PF6− as supporting electrolyte at 100 mV s−1. The potentials are quoted vs. SCE, under our conditions E1/2Fc+/Fc = 0.43 V.d Energy gap between vibrational levels (ν = 0) at the ground and excited state. Determined by taking the wavelength (λ0.05) corresponding to 5% of the maximum intensity of the emission spectrum recorded at 298 K (E0–0 = 1240/λ0.05).e First oxidation potential at excited state. | |||||||
| DTP1 | 415 | 18000 |
594 | 0.85 | −1.50 | 2.48 | −1.49 |
| 334 | 25000 |
496, 594 | |||||
| DTP2 | 457 | 27100 |
685 | 0.68 | −1.45 | 2.13 | −1.45 |
| 364 | 18700 |
542, 685 | |||||
The UV-vis absorption spectra (Fig. 1) showed different profiles depending on the substituted ring of the DTP core.
An intense π–π* band was observed at 334 nm (ε ≈ 25000 M−1 cm−1) in DTP1 followed by another less intense one (ε ≈ 18000 M−1 cm−1) in the visible part (415 nm). Moving the styrene from pyrrole to thiophene on DTP i.e. going from DTP1 to DTP2 induced a large bathochromic effect for both bands. In this case the band in the visible part (457 nm) was the most intense (ε = 27000 M−1 cm−1) signing an enhancement of the ICT process. The dyes were found emissive in fluid dichloromethane solutions. The electrochemical properties have been studied using cyclic voltammetry (Table 1). The first oxidation potential that is directly related to the HOMO level of the dyes has been measured and found at 0.85 V and 0.68 V per SCE for DTP1 and DTP2 respectively. These values indicate that the regeneration of the oxidized dye ground state by the common I3−/I− couple (0.2 V vs. SCE) is thermodynamically favored. The determination of the E0–0 (zero–zero energy) from emission spectra allowed the calculation of the excited state oxidation potential (E*ox = Eox − E0–0). E*ox was found near −1.45 V for both dyes i.e. positioned at much more negative values than the flat band potential of TiO2 (−0.7 V vs. SCE). Thus the injection of the excited electron into the conduction band of the semiconductor is expected to be efficient.
Computational analysis
The absorption spectra of both DTP1 and DTP2 dyes were calculated at Time-Dependent Density Functional Theory (TD-DFT) level. The main features of the experimental absorption spectrum were nicely reproduced (Fig. 2). Indeed the calculated spectra of both dyes are characterized by an intense low-energy transition appearing in the visible. Note that the low energy part of the spectrum is characterized by the presence of only one vertical transition, happening at 403 and 418 nm for DTP1 and DTP2, respectively. Hence TD-DFT results nicely confirm the red-shift of DTP2. Note also that the blue shift compared to the experimental values can also come from the neglecting of dynamical effects and vibrational couplings. | ||
| Fig. 2 Convoluted TD-DFT spectra of dyes. | ||
To characterize the excited states nature, NTOs (natural transitions orbitals)11 were calculated for the dyes in order to get further insight into the electron distribution (Fig. 3). “Occupied” NTO (oNTO) can be seen as the orbital from which the electron is removed during the transition, while “virtual” NTO (vNTO) is the orbital in which the electron is placed in the excited state. For an efficient electron transfer upon solar irradiation, oNTO must be delocalized through the donor moiety while the vNTO has to be located in the anchoring group through the π-bridge.
 | ||
| Fig. 3 NTOs of dyes. | ||
In DTP1, oNTO is mainly delocalized over the pyrrole ring of DTP and the π-conjugated bridge (styryl). The DTP is no more significantly involved in vNTO. For dye DTP2, oNTO is delocalized over the three heteroaromatic rings of the DTP and the π-conjugated bridge while the vNTO is mainly located in the cyanoacrylic group through the π-bridge. The more extended delocalization of the π-system in the DTP2 transition orbitals compared to DTP1 gives also a clear interpretation of the observed red-shift of these dyes.
Compared with the other dye, in DTP2, the accepting cyanoacrylic group is insignificantly implied in the oNTO thus leading to an expected better charge separation. Conversely, and positively in term of charge-injection into the semiconductor surfaces, the vNTO of both dyes is significantly delocalized over the bridging cyanoacrylic group.
As expected, the hexylphenyl substituent of the pyrrole ring of DTP is not involved in the delocalization due to its orthogonality towards the DTP plane. This particular geometry has been chosen to prevent dye molecules aggregation and subsequent potential excited states deactivation.
Photoelectrochemical measurements
DTP1 and DTP2 were chemisorbed onto TiO2 films and the sensitization was monitored by UV-vis spectroscopy (Fig. 4). As shown, a wide absorption of the visible part of the spectrum after grafting is the signature of an efficient sensitization of the semi conducting anode by DTP2. This dye exhibited high absorbance (>3) in the 300–550 nm region that slowly decreased until 700 nm. DTP2 was found much better sunlight harvester than one of the best known ruthenium complex Z907 (cis-bis(isothiocyanato) (2,2′-bipyridyl-4,4′-dicarboxylato) (4,4′-di-nonyl-2′-bipyridyl)ruthenium(II)).12 The sensitization with DTP1 was less efficient with a narrower absorption window (300–480 nm) in agreement with the absorption in solution. | ||
| Fig. 4 Overlay absorption spectra of TiO2 photoanodes modified with the dyes. | ||
Then we turned to the evaluation of the photovoltaic properties and DSSCs were assembled following reported methods using the new dyes. As redox mediator, a mixture of 1-propyl-3-methyl-imidazolium iodide (PMII) (0.6 M), I2 (0.1 M), LiI (0.1 M) in propionitrile containing t-butylpyridine (TBP, 0.5 M) was used. Upon adsorption on the TiO2 surface TBP protects it from recombination with I3− ions while tuning the Fermi level of the semiconductor. For each dye, DSSC were prepared using a transparent (≈7 μm) and an opaque TiO2 anode (≈7 μm) fabricated with commercial P25 nanoparticles.2a
The photovoltaic data are gathered in Table 2 and J–V curves are given in Fig. 5. All measurements have been made under the standard AM 1.5 G irradiation (100 mW cm−2).
| Dye | Jsc (mA cm−2) | Voc (mV) | FF | η% |
|---|---|---|---|---|
| a Measurements performed under AM 1.5 G irradiation (100 mW cm−2), irradiated area: 0.5 cm2 on thin TiO2 layers (6–7 μm). In parentheses: obtained with the opaque TiO2 photoanode. | ||||
| DTP1 | 4.29 (6.55) | 570 (620) | 0.61 (0.61) | 1.49 (2.46) |
| DTP2 | 6.82 (7.60) | 620 (610) | 0.63 (0.60) | 2.68 (2.80) |
| Z907 | 8.57 (8.50) | 680 (640) | 0.66 (0.64) | 3.85 (3.46) |
 | ||
| Fig. 5 J–V curves for DTP1, DTP2 and Z907 using transparent (top) and opaque (bottom) TiO2. | ||
DTP2-based device performed better than those made from DTP1. Using the transparent 6 μm TiO2 layer, respective efficiencies of 1.49 and 2.68% were obtained for DTP1 and DTP2 with associated photocurrents of 4.29 and 6.82 mA cm−2 and open circuit voltages of 570 and 620 mV. Under the same conditions, the efficiency of the Ru-based Z907 was 3.85% and the generated photocurrent was 8.57 mA cm−2. The Fill Factor of the devices was found in the 0.61–0.66 range. With the opaque TiO2 layer, an improved light harvesting efficiency was expected. The consequence was a remarkable gap of the efficiency of the DTP1-based cell from 1.49 to 2.46% this was concomitant to an increase of the photocurrent up to 6.55 mA cm−2 and Voc (620 mV). The effect of the opaque electrode was also beneficial to the DTP2-based device but in a lesser extent since the efficiency obtained was 2.80% instead of 2.68 with the transparent electrode. In this case, the increase of the photocurrent from 6.82 to 7.60 mA cm−2 was not associated with an improvement of the Voc. The opaque electrode was detrimental to the performance of the Z907-based device, essentially due to a lower Voc, resulting in an efficiency of 3.46%, quite close to that observed with the DTP2-based device (2.80%). Note that in this case the photocurrent remained almost constant.
In order to investigate the absorption injection relationship with the new dyes, IPCE measurements have been performed (Fig. 6). As shown, both dyes showed good IPCE maximum values up to 80%. In agreement with the absorption spectra of chemisorbed dyes on TiO2 (Fig. 4), the DTP2-based cell showed red shifted maximum compared with DTP1. On transparent photoanode (Fig. 6 (top)), DTP2 showed a high conversion efficiency (>70%) in the 450–550 region with an onset near 700 nm. The action domain of DTP1 was found narrower with a maximum conversion efficiency (>70%) concentrated in the 450–500 range. The lower photocurrent measured from DTP1-based cell is thus explained by its lower light harvesting efficiency in the red portion of the visible spectrum compared with DTP2. The absorption domain covered by DTP2 was found intermediate between those of DTP1 and Z907. DSSC built from opaque TiO2 generally exhibited lower but broader photoaction spectra, with IPCE maxima in the 60–70% range. The increased light scattering of the opaque substrate resulted in a considerable broadening of the action spectra of the DTP based organic dyes, which is consistent with the improved photocurrents observed in J/V experiments. In particular, compared to the transparent substrate, the action spectrum of the organic dyes undergoes the most significant enhancement in the low energy region of their absorption spectrum, where the extinction coefficient is smaller, that is, in the 600–700 nm and in the 550–650 nm intervals for DTP2 and DTP1 respectively. This effect is less pronounced with Z907, already displaying a good red response, where the broader photoresponse is partly offset by the decreased absolute conversion efficiency, leading to the substantially unaltered Jsc observed under AM 1.5 G illumination (Table 2 and Fig. 5).
 | ||
| Fig. 6 Action spectra for DTP1, DTP2 and Z907 using transparent (top) and opaque (bottom) TiO2. Values corrected for the transmittance of the FTO. | ||
Photophysics
Time-correlated single photon counting (TCSPC) measurements were performed on dye solutions in dichloromethane applying a pulsed 460 nm excitation (see ESI†). The dyes were characterized by singlet excited state lifetimes of 1.0 and 1.8 ns for DTP1 and DTP2 respectively. 355 nm laser excitation (FWHM 7 ns) results in the instrument response limited population of the lowest triplet excited state, which is characterized by a generally weak spectroscopic signature (maximum ΔOD ≈ 3 × 10−3) and displays a lifetime in the range of hundreds ns, (190 ± 10 ns (DTP1) and of 390 ± 20 ns (DTP2)). The triplet state decays according to a mono-exponential function, with R ≈ 0.99 (Fig. 7). As shown, DTP1 (top) and DTP2 (bottom) exhibited different excited state signatures. However, both spectra showed a significant bleaching of the ground state at λ < 520 nm. At longer wavelengths DTP1 showed a broad and featureless absorption (λmax = 650 nm). The maximum of absorption for DTP2 was also found at the same value, but the triplet–triplet absorption showed two narrower bands with distinct maxima at 600 and 650 nm and a third smaller one at 750 nm. | ||
| Fig. 7 Transient absorption spectra following 355 nm ns laser excitation, represented at increasing delays after the laser pulse (t0). DTP1 (top) and DTP2 (bottom). | ||
TA spectroscopy was also applied to dye-chemisorbed transparent TiO2 electrodes immersed in acetonitrile–LiClO4 (0.1 M) solutions (Fig. 8). The oxidized state of the dyes was instantaneously generated by photoinduced charge injection into the conduction band of TiO2 following a 532 nm laser excitation (4.8 mJ per cm2 per pulse). The oxidized state recovery followed a multiexponential kinetics, imposed by recombination with electrons trapped into the TiO2. In the presence the Li+ containing electrolyte, the recovery was incomplete on the thousands of ns time scale. Fig. 8 clearly revealed that the spectra collected for the respective dyes at delays larger than 2.8 μs for DTP1 (top) and 560 ns for DTP2 (bottom) are indeed essentially superimposable. Qualitatively similar recombination kinetics were also found for the reference Z907 dye, where ca. 2/3 of the initial amplitude decays within the first 160 ns and the remaining 1/3 exhibited a very slow kinetic, illustrated by the nearly overlapping spectra recorded at delays >160 ns. Both the characteristic 600–650 nm absorption feature and intense shorter wavelength bleaching were instantaneous for the time resolution of the nanosecond spectrometer apparatus that is consistent with the ultrafast injection reported for dyes having cyano-acrylic binding groups.13
 | ||
| Fig. 8 TA spectra of dyes chemisorbed on transparent TiO2 following 532 nm laser excitation (4.8 mJ per cm2 per pulse). DTP1 (top), DTP2 (bottom). Inset: expanded view of the long wavelength absorption of DTP2. | ||
Interestingly, the oxidized dye absorption spectrum of DTP1-sensitized TiO2 (Fig. 8, top) had a strong analogy with those of the excited state of the dye in solution (Fig. 7, top) with however a significantly increased amplitude of the bleaching at λ < 550 nm compared to the longer wavelength absorption. The same observation was made with DTP2, where the ground state bleaching dominates the 450–600 nm range and is followed by a relatively sharp absorption band peaking at 670 nm (Fig. 8, inset), which resembles the group of bands observed in the excited state of the same dye in solution and located in the 600–670 nm region (Fig. 8, bottom). Thus, by comparison with the TA spectra of the photo-oxidized species on the TiO2 surface, the long wavelength absorption of the excited state of DTP1 and DTP2 dyes can be reasonably assigned to the hole located on the donor group of the D–π–A system. Such species is transiently generated also at the excited state, in agreement with the strong charge transfer character of the lowest excited state evidenced by quantum-mechanics calculations.
The analysis of the recombination kinetics of the DTP-based dyes (Fig. 9), compared with those of Z907, allowed to exclude a faster direct recombination of the oxidized DTP-based sensitizer with photo-injected electrons. Indeed, in the presence of inert electrolyte Li+ (Fig. 9 blue line), the initial part of the decay of the DTP dyes was significantly slower than in the case of Z907, showing a τ2/3 of 4.5, 0.76 μs and 0.155 μs for respectively DTP1, DTP2 and Z907. This behavior was consistent with a hole confinement into the donor part of the dye which should be farther from the semiconductor surface resulting in a smaller electronic coupling an thus reducing the tunneling probability from the TiO2 surface.
 | ||
| Fig. 9 Normalized oxidized dye recovery in the presence of different electrolytes: 0.1 M Li+ (blue); 0.05 M I3− (green); 0.1 M LiI/0.6 PMII (red). DTP1 (top) observed at 600 nm (a). DTP2 (middle) observed at 650 nm (b). Z907 (bottom) observed at 770 nm (c). Decays were fitted according with tri-exponential functions. | ||
In order to evaluate the regeneration efficiency, the electrodes were placed in the presence of electron donating electrolyte consisting of 0.6 PMII and 0.1 M LiI. In this case, the dye recovery was substantially accelerated by electron transfer from I− to the oxidized dye (Fig. 9 red line). τ2/3 was reduced to 0.43, 0.19 and less than 0.04 μs for DTP1, DTP2 and Z907 respectively. However, while the dye recovery was completed within ≈350 ns after the laser pulse for Z907, it was achieved only within ca. 10 μs for DTP2 and incomplete for DTP1 on the time scale of the measurement (10 μs). Thus in the presence of an incomplete recovery in case of DTP1, only an approximate estimate of the regeneration efficiency ηreg could be made according to eqn (1) where τ2/3reg and τ2/3rec are the lifetimes obtained respectively in the presence (donating electrolyte) and in the absence (inert electrolyte) of I−.
 | (1) |
Eqn (1) applied to our data led to regeneration efficiencies of 80% for both Z907 and DTP2 and 91% for DTP1. In this latter case the regeneration efficiency was most probably overestimated due to the incomplete dye recovery. Nevertheless these regeneration efficiencies were in good agreement with the maximum IPCE values close to 80% (IPCE = Φinjηcoll when LHE ≈ 1). Since the electron collection efficiency ηcoll can be identified with ηreg when losses by electron recombination with I3− (the oxidized form of the redox mediator) are negligible, the quantum yield of charge injection Φinj was nearly unitary in the experimental conditions used for all dyes under investigation.
The process of recombination between electrons in TiO2 and I3− can be indirectly probed by TA spectroscopy by monitoring the decay of the dye in the presence of an electrolyte containing exclusively I3− that subtracts electrons from TiO2 according to I3− + 2e− → 3I−. Thus electrons are made unavailable to the direct e−/Dye+ recapture, resulting in an increase of the Dye+ lifetime at the surface. This effect was generally observed for all dyes (see green lines in Fig. 9).
The apparent first order recombination rate constant krecI3− can be calculated by using the relevant τ2/3 parameters according to the simple eqn (2).
 | (2) |
krecI3− of 4.8 × 105, 7.9 × 105 and to 1.9 × 106 s−1 were respectively obtained from eqn (2) for DTP1, DTP2 and Z907. Thus the recombination rate involving I3− on the dyed semiconductor surface should decrease in the order Z907 > DTP2 > DTP1.
In order to corroborate these results, symmetrical thin-layer dummy cells constituted by two identical dyed TiO2 electrodes were assembled. Steady state voltammetry confirmed, albeit only at a qualitative level, that the recombination current varied in the same order Z907 > DTP2 > DTP1 (Fig. 10). It should be noted that the TA measurements address the recombination involving only the TiO2 and not the FTO back contact, given that it is the recovery of the oxidized state of the dye which is directly monitored. Recombination involving the FTO is clearly included in the recombination current recorded in the electrochemical measurement without changing the relative ordering deduced by TA experiments.
 | ||
| Fig. 10 J–V curves in symmetrical dummy cells constituted by dyed photoanodes. Scan rate 2 mV s−1. Electrolyte 0.6 M PMII/0.1 M LiI/0.5 M TBP and 0.1 M I2 in propionitrile. | ||
Conclusions
A new family of donor–π organic dyes based on the DTP electron donor was prepared and characterized under the fundamental aspects concerning sensitization of wide band gap semiconductors. These new dyes generated maximum monochromatic photoconversion efficiencies (IPCE) comparable to that of standard Ru(II) dyes, resulting in maximum IPCEs close to 80%. This is consistent with nearly quantitative charge injection yields, as confirmed by TA measurements revealing the instrument-response limited formation of the dye-cation. Dye regeneration yields were also found ≥80%, whereas the electron recapture rate constant by the I3− was indirectly estimated from the TA kinetics of being, in the best cases about 1/2 of that found with the Z907 dye, in agreement with the dark currents observed in symmetric dummy cells comprised of sensitized TiO2 electrodes. Thus, in the new dyes, the kinetic competition between charge separation and recombination is well suited to achieve an efficient sensitization. The lower efficiency of the DTP dyes with respect to standard Ru(II) dyes, appears to be mainly determined by an insufficient red response of the dyes, as demonstrated by the substantially increased photocurrents observed with opaque TiO2 photoanodes, where the increased light scattering allowed to harvest more effectively photons whose energy is at the onset of the electronic spectrum of the DTP sensitizers.Experimental part
General methods
All reagents were commercially available and used as received. THF was dried using a solvent purification device. DMF was distilled over CaH2. Other solvents were used as purchased. Compounds 1, 28 and 310 were prepared according to literature procedures. Thin-layer chromatography (TLC) was performed on silica gel 60 with fluorescent indicator UV254 (0.2 mm) with UV detection. 1H and 13C NMR spectra were obtained on 250 or 400 MHz spectrometers. 1H and 13C NMR chemical shifts (δ) are reported relative to Me4Si used as an internal standard. Coupling constants are reported in Hertz (Hz). UV-vis spectra were recorded in a 1 cm path length quartz cell on a LAMBDA 1050 (Perkin Elmer), spectrophotometer. Emission and Excitation spectra were obtained on optically diluted solutions by using a Fluoromax-2 (Horiba Jobin Yvon) spectrofluorometer. Cyclic voltammetry was performed on a Radiometer PST006 potentiostat using a conventional three-electrode cell. The saturated calomel electrode (SCE) was separated from the test compartment using a bridge tube. The solutions of studied complexes (0.5 mM) were purged with argon before each measurement. The test solution was DMF containing 0.1 M Bu4NPF6 as supporting electrolyte. The working electrode was a 10 mm Pt wire and the counter-electrode was a 1 cm2 vitreous carbon disc. After the measurement, ferrocene was added as the internal reference for calibration. All potentials were quoted versus SCE. In these conditions the redox potential of the couple Fc+/Fc was found at 0.45 V. In all the experiments the scan rate was 100 mV s−1.
Synthesis of dyes
:4:1) as eluents afforded the title compound DTP2 (58 mg, 73%). δH (250 MHz, DMSO-d6) 0.84 (t, J = 7.3 Hz, 3H), 1.26 (m, 6H), 1.63 (m, 2H), 2.72 (t, J = 7.3 Hz, 2H), 6.58 (d, J = 3.8 Hz, 1H), 6.63 (t, J = 3.8 Hz, 1H), 6.67 (s, 1H), 6.74–6.68 (m, 2H), 6.88 (dd, J = 5.1 and 3.6 Hz, 1H), 7.00 (d, J = 3.9 Hz, 1H), 7.42–7.25 (m, 5H), 7.59 (d, J = 8.2 Hz, 2H), 7.86 (d, J = 8.3 Hz, 2H), 7.94 (s, 1H) ppm. δC (62.5 MHz, DMSO-d6) 13.9, 22.1, 27.8, 30.7, 31.0, 34.7, 109.7, 109.9, 112.2, 119.3, 123.7, 124.2, 124.9, 125.7, 126.5, 127.1, 128.1, 129.6, 129.8, 130.5, 131.7, 132.0, 133.9, 134.1, 135.1, 139.1, 139.9, 144.4, 146.9, 163.6 ppm. HRMS calcd for C36H31N2O2S2 [M + H]+ 587.1832. Found: 587.1837.Preparation of DSSCs and photoelectrochemical measurements
The nanocrystalline TiO2 photoanodes were prepared by depositing either the transparent TiO2 paste (Dyesol DS 18N-RT) or the opaque TiO2 P25 paste2a onto transparent conducting FTO glass (Pilkington TEC 8 8 Ω per Square, 2.3 mm in thickness) according to the well-known “scotch tape” method. The thin films were allowed to dry at room temperature for 20 min and annealed by using a temperature program consisting in first ramp to 400 °C (18.8 °C min−1), followed by heating at 400 °C (20 min). A second ramp (10 °C min−1) brought the temperature at 500 °C which was maintained for additional 10 min. After this stage, the electrodes were finally allowed to cool slowly at room temperature. This method allows avoiding fracturing of the film during sintering. TiCl4 treatment was performed by soaking the photoanodes in 0.4 M TiCl4 at room temperature for 12 hours, following by sintering at 450 °C for 30 minutes. The thickness of the TiO2 films was approximately 7 μm. The still hot electrodes were immersed in the dye solution in dichloromethane and stored in a refrigerator at ca. 4 °C overnight, after which the absorption was deemed complete resulting in electrodes with maximum optical densities close to 3. Saturated organic dye solutions were prepared by dissolving the selected dyes in dichloromethane. The solutions were sonicated and filtered to remove suspended undissolved dye. Platinum coated counter electrodes were obtained by screenprinting of printable platinum paste (Chimet) on FTO glass. Parafilm® sealed cells were built by pressing the sensitized photoanode against a counter electrode equipped with a parafilm® frame used to confine the liquid electrolyte inside the cell. The thickness of the liquid layer corresponded roughly to the thickness of the frame borders (≅100 μm). In this configuration the cell was stable towards solvent evaporation and leaking for ca. 1 week. The irradiated surface was 0.25 cm2. Photoelectrochemical measurements were performed with a custom made apparatus built by Dyers under the light output provided by an Oriel 68806 xenon lamp. Illumination of the cells occurred through an optical fiber whose distance from the top electrode (photoanode) of the cell was adjusted in order obtain an incident irradiance of 100 mW cm−2. Data were collected and elaborated on a PC controlled by Lab View 2011 software.Photophysical measurements
Time Correlated Single Photon Counting (TCSPC) measurements were performed with a previously described apparatus8 by exciting optically diluted dye solutions at 460 nm. Differential Transient Absorption Spectra (TAS) in both solution and on sensitized TiO2 thin films were acquired with a time resolved spectrometer employing a Q-switched Nd-YaG laser as a nanosecond (FWHM = 7 ns) excitation source. The excitation of the photoanodes was achieved with the 2nd harmonic (532 nm) of the laser, which was defocused with a plano concave lens and attenuated with a 50% T neutral density filter to ca. 4.8 mJ per cm2 per pulse. The probe beam, generated by a 150 W Xe lamp, attenuated by combining a 420 nm cut off filter and 25% T neutral density filter was passed through the thin film oriented at 45° and was focused into the slit of an Acton Spectra Pro 2300i monochromator (150 lines per mm) equipped with a Hamamatsu R3896 phototube. A 532 nm notch filter prevented scattered laser light to reach the detector. Oscillographic traces were acquired on a LeCroy 9360 oscilloscope, averaged over 30 laser shots and transferred to a PC. A custom made Lab View program controlled laser firing, pulse synchronisation and data acquisition. The strong absorption of the organic dyes below 450 nm and the presence of the 420 nm cut off filter, necessary for avoiding direct TiO2 excitation, prevented the exploration of the visible spectrum below 450 nm. TAS in solution were obtained by exciting at 355 nm with the same experimental procedure, except that that the optics for laser and probe beam attenuation were removed.Computations
All quantum chemistry calculations have been performed by using Gaussian 09 suite. Ground state equilibrium geometry has been calculated at DFT level using the double-zeta plus polarization 6-31G(d) basis set and the long range-corrected CAM-B3LYP functional. Excited states have been calculated using Time Dependent DFT (TD-DFT) as vertical excitations from ground state equilibrium geometry. For the TD-DFT calculations we used 6-31+G(d,p) basis set and the wB97XD functional. Solvent effects were taken into account using polarizable continuum methods (PCM). The levels of theory for geometry optimization and excited state calculations were chosen since this combination was the one giving the best agreement between computed and experimental absorption spectra. Excited states analysis in terms of NTOs was performed by using a locally produced and free downloadable code Nancy EX (see http://www.nancyex.sourceforge.net/).Acknowledgements
The authors would like to thank the CNRS, the French Ministry of Research and STDF-AIRD program for support. W.S thanks the NRC (Cairo) as well as French government for a grant. Funding from Erasmus Program (A.A.) and PRIN 2011 (UNIFE) is also gratefully acknowledged.Notes and references
- (a) B. O'Regan and M. Gräzel, Nature, 1991, 353, 737 CrossRef; (b) A. Hagfeldt and M. Grätzel, Chem. Rev., 1995, 95, 49 CrossRef CAS; (c) R. Argazzi, C. A. Bignozzi, G. M. Hasselmann and G. J. Meyer, Inorg. Chem., 1998, 27, 4533 CrossRef PubMed; (d) A. E. Curtright and J. K. McCusker, Inorg. Chem., 1999, 35, 7032 Search PubMed; (e) M. I. J. Polson, N. J. Taylor and G. S. Hanan, Chem. Commun., 2002, 1356 RSC; (f) P. Wang, C. Klein, R. Humphry-Baker, S. M. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 808 CrossRef CAS PubMed; (g) S.-L. Li, K.-J. Jiang, K.-F. Shao and L.-M. Yang, Chem. Commun., 2006, 26, 2792 RSC; (h) N. Robertson, Angew. Chem., Int. Ed., 2006, 45, 2338 CrossRef CAS PubMed; (i) M. K. Nazeeruddin, S. Zakeerudin, J.-J. Lagref, P. Liska, P. Comte, C. Barolo, G. Viscardi, K. Schenk and M. Graetzel, Coord. Chem. Rev., 2004, 248, 1317 CrossRef CAS PubMed; (j) For a recent book see: Dye-Sensitized Solar Cells, ed. K. Kalyanasundaram, EPFL Press, Lausanne, 2010 Search PubMed.
- (a) M. K. Nazeeruddin, A. Kay, L. Rodicio, R. Humphry-Baker, E. Muller, P. Liska, N. Vlachopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382 CrossRef CAS; (b) M. K. Nazeeruddin, P. Pechy, T. Renouard, S. M. Zakeeruddin, R. Humphry-Baker, P. Comte, P. Liska, L. Cevey, E. Costa, V. Shklover, L. Spiccia, G. B. Deacon, C. A. Bignozzi and M. Grätzel, J. Am. Chem. Soc., 2001, 123, 1613 CrossRef CAS PubMed.
- (a) A. Mishra, M. K. R. Fischer and P. Bauërle, Angew. Chem., Int. Ed., 2009, 48, 2474 CrossRef CAS PubMed; (b) J. N. Clifford, E. Martinez-Ferrero, A. Viterisi and E. Palomares, Chem. Soc. Rev., 2011, 40, 1635 RSC.
- (a) Y. Ooyama and Y. Harima, Eur. J. Org. Chem., 2009, 2903 CrossRef CAS; (b) A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Petterson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed.
- (a) M. Fujii, T. Nishinaga and M. Iyoda, Tetrahedron Lett., 2009, 50, 555 CrossRef CAS PubMed; (b) M. Moreno Oliva, T. M. Pappenfus, J. H. Melby, K. M. Schwaderer, J. C. Johnson, K. A. McGee, D. A. da Silva Filho, J.-L. Bredas, J. Casado and J. T. Lopez Navarrete, Chem.–Eur. J., 2010, 16, 6866 CrossRef PubMed.
- (a) G. Zotti, S. Martina, G. Wegner and A.-D. Schlüter, Adv. Mater., 1992, 4, 798 CrossRef CAS; (b) R. E. Niziurski-Mann, C. Scordilis-Kelley, T. L. Liu, M. P. Cava and R. T. Carlin, J. Am. Chem. Soc., 1993, 115, 887 CrossRef CAS; (c) P. Audebert, S. Sadki, F. Miomandre, P. Hapiot and K. Chane-Ching, New J. Chem., 2003, 27, 798 RSC; (d) T. M. Pappenfus, B. J. Hermanson, T. J. Helland, G. G. W. Lee, S. M. Drew, K. R. Mann, K. A. McGee and S. C. Rasmussen, Org. Lett., 2008, 10, 1553–1556 CrossRef CAS PubMed.
- (a) S. Yanagida, Y. Yu and K. Manseki, Acc. Chem. Res., 2009, 42, 1827 CrossRef CAS PubMed; (b) C.-Y. Chen, S.-J. Wu, C.-G. Wu, J.-G. Chen and K.-C. Ho, Angew. Chem., Int. Ed., 2006, 45, 5822 CrossRef CAS PubMed.
- (a) S. Noureen, S. Caramori, A. Monari, X. Assfeld, R. Argazzi, C. A. Bignozzi, M. Beley and P. C. Gros, Dalton Trans., 2012, 41, 4833 RSC; (b) S. Noureen, R. Argazzi, A. Monari, X. Assfeld, M. Beley, C. A. Bignozzi, S. Caramori and P. C. Gros, Dyes Pigm., 2014, 101, 318 CrossRef CAS PubMed.
- (a) V. Tamilavan, N. Cho, C. Kim, J. Ko and M. Ho Hyun, Tetrahedron, 2012, 68, 5890 CrossRef CAS PubMed; (b) V. Tamilavan, A.-Y. Kim, H.-B. Kim, M. Kang and M. Ho Hyun, Tetrahedron, 2014, 70, 371 CrossRef CAS PubMed.
- T. Nishizawa, H. K. Lim, K. Tajima and K. Hashimoto, J. Am. Chem. Soc., 2009, 131, 2464 CrossRef CAS PubMed.
- (a) J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC; (b) A. Monari, T. Very, J.-L. Rivail and X. Assfeld, Comput. Chem. Graph Theory, 2012, 990, 119 CrossRef CAS PubMed.
- S. M. Zakeeruddin, M. K. Nazeeruddin, R. Humphry-Baker, P. Pechy Quagliotto, C. Barolo, G. Viscardi and M. Graëtzel, Langmuir, 2002, 18, 952 CrossRef CAS.
- A. K. Biswas, S. Barik, A. Sen, A. Das and B. Ganguly, J. Phys. Chem. C, 2014, 118, 20763 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of dyes and additional photophysical data. See DOI: 10.1039/c4ra10342d |
| ‡ Current address: Department of Inorganic Chemistry National Research Center, Dokki, Cairo 12622, Egypt. |
| This journal is © The Royal Society of Chemistry 2015 |