
Synthesis, characterization and enhanced bactericidal action of a chitosan supported core–shell copper–silver nanoparticle composite†
Abstract
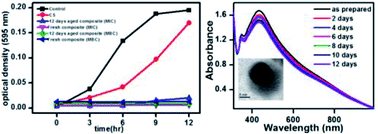
A simple two-step seed-mediated method has been developed to synthesize chitosan (CS)-supported core–shell metal nanoparticles (NPs) in aqueous solution. CS-supported copper NPs (CS–Cu NPs), when treated with silver nitrate (AgNO3) solution, resulted in Ag shells on Cu NP cores in the composite. The Cu@Ag NPs in the CS composite were characterized and found to be spherical in shape with an average particle size of 14.0 ± 3.4 nm. The antibacterial efficacy of the CS–Cu@Ag NP composite was evaluated on Gram-negative Escherichia coli and Gram-positive Bacillus cereus bacteria using turbidity measurement as well as flow-cytometry. Results demonstrated enhanced bactericidal activity of the composite. Analytical techniques, used to elucidate the mode of bactericidal action, revealed that the superior antibacterial activity of the CS–Cu@Ag NP composite was due to synergistic effects of CS and Cu@Ag NPs. The proposed mode of antibacterial action involves the electrostatic binding of CS to the bacterial cell wall and simultaneous interaction of Cu@Ag NPs with membrane proteins leading to perforation of the cell membrane and release of intracellular material.
 Please wait while we load your content...
Please wait while we load your content...

