
A facile synthetic approach for SiO2@Co3O4 core–shell nanorattles with enhanced peroxidase-like activity†
Syam Kandula and
Pethaiyan Jeevanandam*
Department of Chemistry, Indian Institute of Technology Roorkee, Roorkee-247667, India. E-mail: jeevafcy@iitr.ac.in; Fax: +91-1332-273560; Tel: +91-1332-285444
First published on 11th December 2014
Abstract
SiO2@Co3O4 core–shell nanorattles with different Co3O4 shell thicknesses have been successfully synthesized by the calcination of SiO2@α-Co(OH)2 at 500 °C. The synthetic approach is facile, economical, and requires no surface modification. The synthesized materials were thoroughly characterized using powder X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FT-IR), thermal gravimetric analysis (TGA), Brunauer–Emmett–Teller (BET) analysis, field emission scanning electron microscopy (FE-SEM), transmission electron microscopy (TEM), and diffuse reflectance spectroscopy (DRS). SEM analysis indicates a hierarchical core–shell morphology for SiO2@Co3O4 and the TEM results indicate the core–shell nanorattle morphology. Diffuse reflectance spectroscopy studies indicate that the SiO2@Co3O4 core–shell nanorattles show two absorption bands in the range 420–450 nm and 700–750 nm related to ligand to metal charge transfer transitions (O2− → Co2+ and O2− → Co3+). The SiO2@Co3O4 core–shell nanorattles act as an artificial peroxidase enzyme mimic with enhanced intrinsic peroxidase-like activity compared to pure Co3O4 nanoparticles and horseradish peroxidase (HRP), a natural enzyme. The SiO2@Co3O4 core–shell nanorattles show higher kcat and kcat/Km values compared to pure Co3O4 and HRP indicating their applicability as an artificial enzyme mimic in biomedicine and bio-sensing.
1. Introduction
In recent times, scientists are paying increasing attention towards the use of metal oxide nanoparticles in various biological applications due to their versatility, sensitivity, stability, etc.1–6 Enzymes are typically proteins that catalyze bio-chemical reactions in the cellular metabolic processes and they enhance the rate of the reactions.7,8 Horseradish peroxidase (HRP) is a natural peroxidase enzyme which is extracted from plants and it catalyzes decomposition of peroxides and oxidation of various substrates.9 HRP is expensive, difficult to store, and it easily becomes inactive. Scientists have an immense interest in developing artificial enzymes (mimics) due to the inherent drawbacks of natural enzymes; they have a tendency to undergo denaturation under environmental conditions with low stability, and low redox activity.7,10 Nanomaterials based enzyme mimics have the advantage of higher stability under harsh conditions, low cost, tunable catalytic activity and they can be potentially useful in different fields of biology.Silica is chemically inert, stable at elevated temperatures and optically transparent. It is useful in many areas such as glass industries, ceramics, semiconductors, photovoltaic cells and it is also used as catalytic support.11–13 Co3O4 is an important p-type antiferromagnetic semiconductor with a normal spinel structure.14 It is used in various applications such as anode materials in lithium ion rechargeable batteries,15 sensors,16 electrochemical devices,17 solar energy absorbers,18 supercapacitors,19 water oxidation,20 and heterogeneous catalysis.21
In recent years, research is focused on core–shell nanoparticles with yolk–shell or nanorattle morphology. Nanorattles refer to hollow shells with solid cores with interstitial hollow space between the core and the shell. They have a large number of applications in bio-sensing, energy storage, catalysis and biomedicine.22–25 Nanorattles exhibit interesting characteristics which are useful in multi-functional areas. One of the most explored applications of these materials is their use as nanocatalysts. Up to now, several methods have been reported for the synthesis of nanorattles and they include hydrothermal synthesis, selective etching, template-route, annealing, and Ostwald ripening.23–28 Tan et al. have reported the synthesis of Au–Pd@SiO2 nanorattles useful in Suzuki cross-coupling reaction.23 Yang et al. have reported the preparation of Fe3O4@air@poly(vinylsilane) nanorattles and they have used them as contrast agents.24 Zhang et al. have reported the preparation of Au@polyanline yolk–shell nanorattles and have used them as flexible memory device.25 Xu et al. have reported the synthesis of Fe3O4@CuSiO3 nanorattles and have investigated their microwave absorption properties.29 In some cases, Co3O4 has been used as the core material and SiO2 has been used as the shell.22,30–32 Zong et al. have reported the confined growth of metal oxide nanocrystals on mesoporous silica spheres which act as the core material.33
Very less attention has been paid towards the use of metal oxide nanoparticles in biological applications. For example, Mu et al. have reported the peroxidase-like activity for Co3O4 nanoparticles and, recently, they have also studied the effect of crystal plane of Co3O4 on the peroxidase-like activity.10,34 Gao et al. have reported that Fe3O4 nanoparticles possess intrinsic peroxidase-like activity.35 In addition to Fe3O4 and Co3O4 nanoparticles, other systems such as graphene supported Au–Pd bimetallic core–shell nanoparticles,36 Au nanoparticles on citrate-functionalized graphene sheets,37 Co–Al layered double hydroxides,38 porous platinum nanotubes,39 CuS–graphene nanosheets,40 and Fe3O4 nanoparticles loaded on graphene oxide-dispersed carbon nanotubes41 have been explored for the intrinsic peroxidase-like activity. Peroxidase-like activity has also been demonstrated in Co3O4 nanoparticles coupled with carbon nitride nanotubes and reduced graphene oxide.42,43 In the present study, SiO2@Co3O4 core–shell nanorattles with uniform Co3O4 porous shell over the silica core have been successfully synthesized using a self-template route by the calcination of SiO2@α-Co(OH)2 at 500 °C. The synthesized materials were thoroughly characterized before examining the intrinsic peroxidase-like activity using 3,3′,5,5′-tetramethyl benzidine (TMB) as the substrate.
2. Experimental
2.1 Reagents
Tetraethyl orthosilicate (98%, ACROS®), ethanol (99.9%, AR), ammonia solution (25%, Rankem, AR), cobaltous nitrate hexahydrate (98%, Rankem, LR), urea (99.5%, Rankem, AR) and hydrogen peroxide (30%, Rankem, AR) were used as received without further purification. The peroxidase substrate, 3,3′,5,5′-tetramethyl benzidine (TMB), was received from Spectrochem chemicals, India.2.2 Synthesis
The SiO2@Co3O4 core–shell nanorattles were synthesized in two steps and the procedure is as follows.2.3 Characterization
Structural analysis of the synthesized samples was carried out using powder X-ray diffraction on a Brucker AXS D8 diffractometer over the 2θ range of 5–85° and the scan rate was 1° min−1. Copper was used as the target (Cu-Kα; λ = 1.5406 Å). FT-IR spectral analysis of the samples was carried out on a Thermo Nicolet Nexus FT-IR spectrometer in the range 400–4000 cm−1 using KBr pellet method. Thermogravimetric analysis patterns of the as prepared samples were recorded on a EXSTAR TG/DTA 6300 instrument, under air, over the temperature range 25–1000 °C and the heating rate was 10° min−1. Morphological studies of the samples were carried out using a FEI Quanta 200F field emission scanning electron microscope (FE-SEM) operating at 15 kV. Elemental analysis was carried out using an energy dispersive X-ray analysis facility coupled with the FE-SEM instrument. Transmission electron microscopy (TEM) images and selected area electron diffraction (SAED) patterns were obtained using a FEI TECNAI G2 transmission electron microscope. For the TEM analysis, the sample powders were dispersed in ethanol and sonicated for about 15 min. Then, one drop each of the suspensions was put on a carbon coated copper grid and allowed to dry in air. Diffuse reflectance spectroscopy (DRS) analysis was carried out on a Shimadzu UV-2450 UV-Visible spectrophotometer using the powder samples in the wavelength range 360–800 nm and BaSO4 (Aldrich) was used as the reference. Specific surface area of the SiO2@Co3O4 samples was obtained by the Brunauer–Emmett–Teller (BET) method using N2 adsorption at 77 K on a Nova 2200e Quantachrome instrument.2.4 Peroxidase-like redox activity and steady state kinetic analysis
To investigate the peroxidase-like redox catalytic activity of the SiO2@Co3O4 core–shell nanorattles, the oxidation of 3,3′,5,5′-tetramethyl benzidine (TMB) was examined in the presence of H2O2. The reaction was studied at room temperature in acetic acid–acetate buffer containing SiO2@Co3O4 nanorattles as the catalyst. First, 30 μg of SiO2@Co3O4 catalyst was dispersed in 3 mL of 0.1 M acetate buffer and sonicated for 3 minutes. Then, 300 μL of 3 mM DMSO solution of TMB and 31 μL of 30% H2O2 (100 mM) were added and the absorbance values at λmax = 652 nm were recorded upto 5 minutes in time course mode on a Shimadzu UV-2450 UV-Vis spectrophotometer. Kinetic studies were carried out using the same instrument. While keeping the TMB concentration constant, kinetic analysis was performed by varying the amount of the SiO2@Co3O4 catalyst (5–60 μg per 3 mL), pH of the buffer (2–12), temperature (20–70 °C), and H2O2 concentration (5–200 mM). Kinetic studies were also carried out under the following conditions; 30 μg catalyst per 3 mL, pH = 5, temperature = 30 °C and TMB concentration = 0.015–0.4 mM while keeping H2O2 concentration as 100 mM. In another kinetic experiment, while keeping the concentration of TMB as 0.3 mM, the concentration of H2O2 was varied (20–200 mM). The apparent rate constant values (kapp) were evaluated from the absorbance data.9,10 The Michaelis–Menten constant (Km) and the maximal reaction velocity (Vmax) were determined from the Lineweaver–Burk double reciprocal plots. The Michaelis–Menten equation can be expressed as| 1/ν = (Km/Vmax)(1/[S]) + 1/Vmax |
3. Results and discussion
3.1 Material characterization
Fig. 1(a) shows the XRD patterns of as prepared SiO2, α-Co(OH)2, and SiO2@α-Co(OH)2. Silica is amorphous. All the XRD patterns show raising background which is attributed to X-ray fluorescence since Cu-Kα was used as the X-ray source during the measurements. The α-Co(OH)2 shows reflections at 12.18°, 24.37°, 33.11°, 38.08°, 45.98°, 52.05°, and 59.71° which are attributed to (003), (006), (012), (015), (018), (100), and (110) crystal planes (JCPDS file no. 46-0605), respectively.45,46 The first two planes in the XRD pattern of α-Co(OH)2 are related to d-spacing between the intercalated layers (d(003) = 2d(006)).47 The XRD patterns of SiO2@α-Co(OH)2 samples show broad peaks of (003), (006), and (110) planes of α-Co(OH)2 with low crystallinity.48,49 The interplanar spacings of α-Co(OH)2 and SiO2@α-Co(OH)2 samples was calculated using the (003) plane and the value is ca. 7.2 Å. | ||
| Fig. 1 (a) XRD patterns of (i) as prepared SiO2, (ii–iv) SiO2@α-Co(OH)2 samples, and (v) α-Co(OH)2. (b) XRD patterns of (i) SiO2, (ii–iv) SiO2@Co3O4 (S1, S2, and S3), and (v) Co3O4. All the samples were calcined at 500 °C. | ||
Fig. 1b shows the XRD patterns of SiO2, Co3O4 and SiO2@Co3O4 samples (S1, S2, and S3) after calcination at 500 °C. Pure Co3O4 shows reflections at 19.09°, 31.26°, 36.89°, 38.47°, 44.77°, 59.19°, 65.27°, 74.26°, and 78.80° corresponding to (111), (220), (311), (222), (400), (511), (440), (620), and (622) planes of cubic Co3O4 (JCPDS file no. 42-1467), respectively. Since the major component in the core–shell nanorattles is SiO2 which is amorphous, the XRD patterns for SiO2@α-Co(OH)2 as well as SiO2@Co3O4 core–shell nanorattles (obtained on calcination) indicate poorly crystallinity. The XRD patterns for α-Co(OH)2 (Fig. 1a(v)) and pure Co3O4 (Fig. 1b(v)), however, show that these samples are more crystalline compared to the other samples. Calcination does not improve the crystallinity since it only converts α-Co(OH)2 present on SiO2 to Co3O4 and Co3O4 exists as small nanoparticles (as evidenced by the TEM results). XRD results indicate the formation of α-Co(OH)2 on SiO2 spheres. On calcination at 500 °C, the formation of Co3O4 nanoparticles on SiO2 is evident.
FT-IR spectra were recorded for as prepared SiO2, α-Co(OH)2, and SiO2@α-Co(OH)2 (Fig. 2a). The IR spectra for calcined SiO2, Co3O4, SiO2@Co3O4 (S1, S2, and S3) samples are shown in Fig. 2b. All the samples show two bands at about 3430 cm−1 and 1630 cm−1 assigned to stretching and bending vibrations of –OH groups of surface physisorbed water molecules. All the samples except the α-Co(OH)2 and Co3O4 show bands at about 1100 cm−1, 800 cm−1, and 475 cm−1 attributed to asymmetric, symmetric stretching, and bending vibration modes of Si–O–Si. Pure SiO2 shows a characteristic band at 950 cm−1 due to Si–OH group.44 The SiO2@α-Co(OH)2 samples and α-Co(OH)2 show IR bands at 2235 cm−1, and 1387 cm−1, attributed to the stretching frequencies of NCO−, and NO3−, respectively. This indicates the presence of NCO− and NO3− in between the layers.47,50 All the samples except SiO2 show a band at 1023 cm−1 attributed to the deformation mode of –OH group.51 The α-Co(OH)2 and SiO2@α-Co(OH)2 samples show a band at about 660 cm−1 attributed to Co–OH vibration. Pure Co3O4 shows two characteristic bands attributed to Co(II)–O stretching when Co2+ ions are in tetrahedral coordination and Co(III)–O stretching when Co3+ ions are in octahedral coordination;50 SiO2@Co3O4 (S1) shows IR bands at 667 cm−1 and 560 cm−1, sample S2 shows bands at 667 cm−1 and 576 cm−1, and sample S3 shows bands at 664 cm−1 and 568 cm−1. In summary, FT-IR results show the evidence for the presence of NCO− and NO3− ions in α-Co(OH)2, and SiO2@α-Co(OH)2 and the presence of Co3O4 in the SiO2@Co3O4 samples.
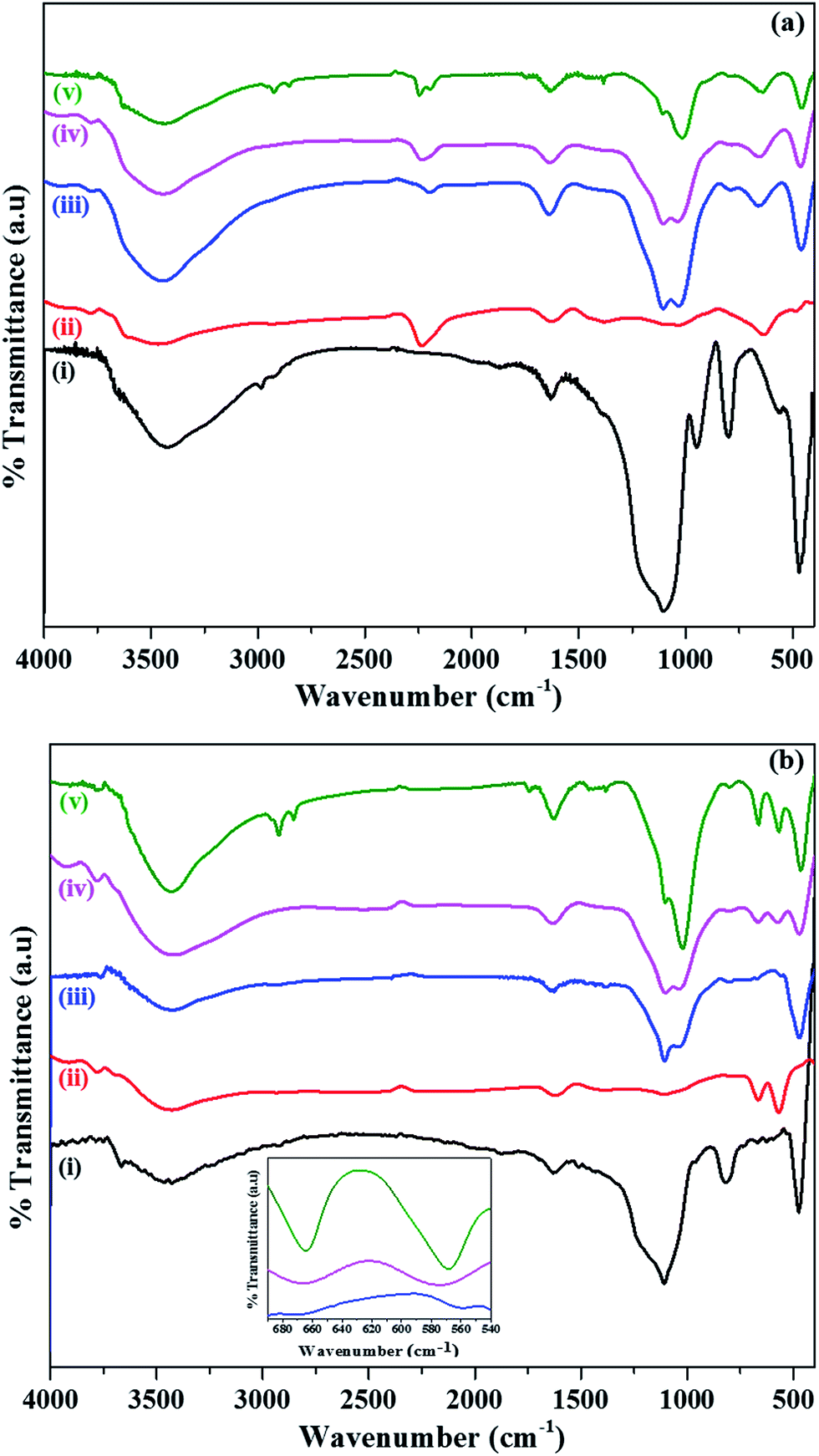 | ||
| Fig. 2 (a) FT-IR spectra of (i) as prepared SiO2, (ii) α-Co(OH)2, (iii–v) SiO2@α-Co(OH)2 and (b) FT-IR spectra of (i) SiO2, (ii) Co3O4, and (iii–v) SiO2@Co3O4 (S1, S2, and S3) samples after calcination at 500 °C. The inset shows the magnified view of IR bands for SiO2@Co3O4 samples (S1, S2, and S3). | ||
The thermogravimetric analysis (TGA) patterns of as prepared SiO2, α-Co(OH)2, and SiO2@α-Co(OH)2 samples are shown in Fig. 3. The as prepared SiO2 shows a total weight loss of 12.5% upto 1000 °C with two weight loss steps. The first step from 35 °C to 170 °C (weight loss = 5%) is attributed to the loss of physisorbed water molecules. The second weight loss from 350 °C to 660 °C (wt loss = ∼7.5%) is attributed to the dehydroxylation of surface hydroxyl groups.52 The α-Co(OH)2 shows a total weight loss of about 35% upto 1000 °C in three steps. The weight loss upto 150 °C (∼2.5%) is related to the loss of surface physisorbed water molecules. The major weight loss (∼28.3%) observed between 220 and 400 °C is attributed to the loss of intercalated water molecules, anions, and dehydroxylation of α-Co(OH)2 accompanied by its conversion to spinel Co3O4.53 Another weight loss (∼4.2%) between 940 and 985 °C is associated with the thermal decomposition of Co3O4 to CoO.54 The SiO2@α-Co(OH)2 samples (S1, S2 and S3) show weight loss features due to both SiO2 and α-Co(OH)2 and the overall weight loss values are 25.5%, 25.2% and 19.5%, respectively. The SiO2@α-Co(OH)2 samples (S1 and S2) show major weight loss (∼12.5%) between 30 and 150 °C, while SiO2@α-Co(OH)2 sample (S3) shows a weight loss of about 4.5% in the same temperature range. Sample S3 (SiO2@α-Co(OH)2 prepared using 15 mmol cobalt nitrate) shows different weight loss features compared to S1 and S2 (SiO2@α-Co(OH)2 samples, prepared using 5 mmol and 10 mmol cobalt nitrate, respectively). It was found from SEM studies (see Fig. 4e) that sample S3 before calcination consists of SiO2@α-Co(OH)2 as well as free α-Co(OH)2 particles. Sample S3 (SiO2@α-Co(OH)2) shows less weight loss below 150 °C (∼4.5%) similar to α-Co(OH)2 (∼2.5%). The low weight loss below 150 °C in these samples (α-Co(OH)2 and S3) is attributed to strong interaction between the surface hydroxyl groups which delay the dehydroxylation process.55,56 The low weight loss at temperature below 150 °C leads to an overall low weight loss in S3. TGA results indicate that the complete formation of spinel Co3O4 occurs at about 500 °C by the loss of intercalated water molecules, anions and dehydroxylation of surface hydroxyl groups of α-Co(OH)2.
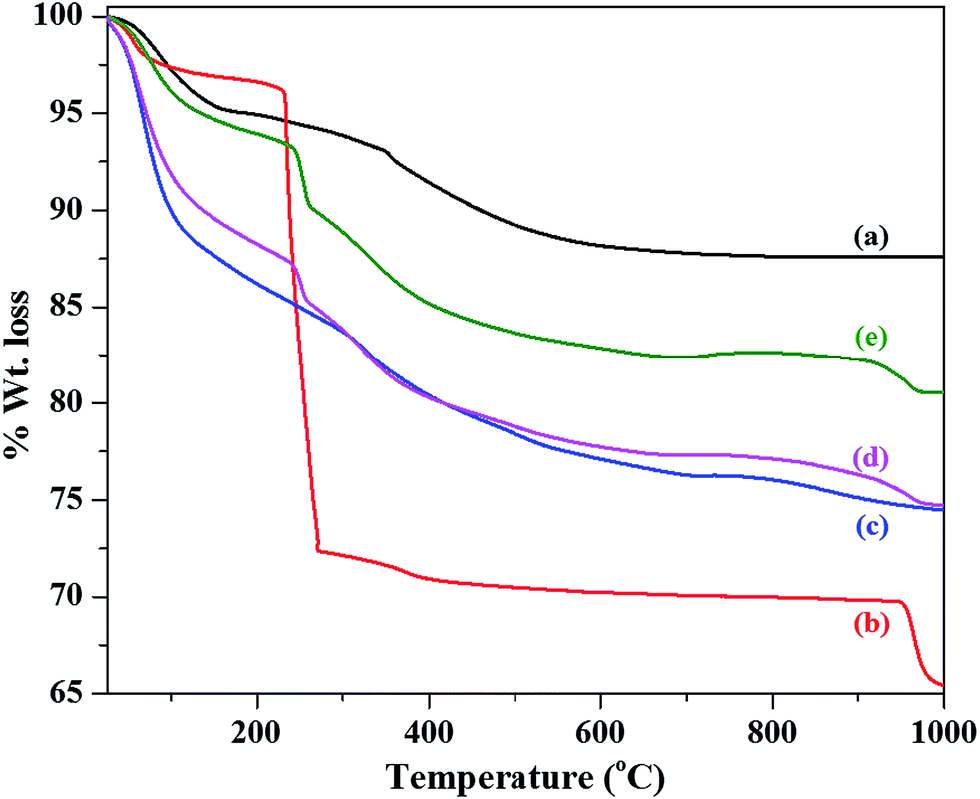 | ||
| Fig. 3 TGA patterns of (a) as prepared SiO2, (b) α-Co(OH)2, and (c–e) SiO2@α-Co(OH)2 samples. | ||
 | ||
| Fig. 4 FE-SEM images of (a) as prepared SiO2 (b) α-Co(OH)2, and (c–e) SiO2@α-Co(OH)2 samples. | ||
Morphological studies of the synthesized samples were first investigated using FE-SEM. The FE-SEM images of SiO2, α-Co(OH)2, and SiO2@α-Co(OH)2 samples (all the samples before calcination) are shown in Fig. 4a–e. Pure silica shows uniform spherical particles and the diameter of the spheres is about 300 ± 20 nm; the average diameter of particles and errors were calculated using about ten particles. α-Co(OH)2 consists of sheets and clusters. The SiO2@α-Co(OH)2 (S1 and S2) show that the silica spheres are completely covered with α-Co(OH)2 sheets (petals) to form a flower-like morphology. But in the case of SiO2@α-Co(OH)2 sample (S3), the SiO2 spheres are covered with α-Co(OH)2 sheets and some uncoated particles of α-Co(OH)2 can also be noticed. The FE-SEM images of Co3O4 and SiO2@Co3O4 samples (i.e. after calcination) are shown in Fig. 5(a–d). Pure Co3O4 shows particles with petals-like morphology. The SiO2@Co3O4 samples (S1 & S2) show uniform shell of Co3O4 over the silica core but sample S3 shows coating of Co3O4 shell on the SiO2 spheres with some extra uncoated particulates of Co3O4. Based on UV-Vis transmittance measurements, it was found that the dispersion of the core–shell particles in water is stable for at least 6 h. It was also found that the powder obtained after drying the particles can be redispersed effectively.
 | ||
| Fig. 5 FE-SEM images of (a) Co3O4, and (b–d) SiO2@Co3O4 samples (S1, S2, and S3). All the samples were calcined at 500 °C. | ||
The elemental composition of the SiO2@α-Co(OH)2 and SiO2@Co3O4 core–shell nanoparticles (S1, S2, and S3) was determined using EDX analysis (Table 1). The EDX results confirm the presence of silicon, oxygen and cobalt in the SiO2@Co3O4 core–shell nanorattles as well as the SiO2@α-Co(OH)2 samples. Increase in the weight percent of cobalt in the core–shell samples is observed according to the concentration of cobaltous nitrate used during the synthesis of SiO2@α-Co(OH)2 samples (5 mM, 10 mM, and 15 mM).
| Sample name | Weight percent | ||
|---|---|---|---|
| Si | Co | O | |
| a The concentrations of Co(NO3)2·6H2O used during the preparation of SiO2@α-Co(OH)2 samples S1, S2 and S3 were 5 mM, 10 mM and 15 mM, respectively. | |||
| SiO2 | 28.0 ± 1.4 | — | 32.5 ± 3.5 |
| α-Co(OH)2 | — | 93.4 ± 1.5 | 6.6 ± 1.5 |
| SiO2@α-Co(OH)2 (S1)a | 15.2 ± 1.9 | 17.6 ± 3.1 | 37.4 ± 2.3 |
| SiO2@α-Co(OH)2 (S2)a | 13.4 ± 0.2 | 21.2 ± 2.6 | 35.1 ± 1.2 |
| SiO2@α-Co(OH)2 (S3)a | 10.9 ± 0.5 | 23.6 ± 1.5 | 38.9 ± 1.3 |
| Co3O4 | — | 83.6 ± 0.7 | 16.4 ± 0.3 |
| SiO2@Co3O4 (S1) | 11.1 ± 0.6 | 14.0 ± 0.5 | 37.9 ± 0.7 |
| SiO2@Co3O4 (S2) | 11.4 ± 0.2 | 16.0 ± 1.0 | 34.8 ± 0.1 |
| SiO2@Co3O4 (S3) | 9.7 ± 1.1 | 27.9 ± 0.3 | 30.3 ± 0.2 |
The TEM images of SiO2, Co3O4 and SiO2@Co3O4 core–shell samples (S1 & S2) are shown in Fig. 6a. TEM analysis was not carried out for the SiO2@Co3O4 sample S3 since the SEM analysis on this sample indicated the presence of uncoated α-Co(OH)2 particles in the SiO2@α-Co(OH)2 sample (see Fig. 4e). SiO2 shows uniform spherical particles with a diameter of about 300 ± 10 nm. The TEM image indicates the nanosized nature of Co3O4 particles (13.7 ± 2.1 nm). The TEM images for the SiO2@Co3O4 samples (S1 & S2) show the core–shell morphology with a void space between the SiO2 core and the Co3O4 shell. These type of structures are referred to as “nanorattles”.24,29 In addition to the nanorattle morphology, the SiO2@Co3O4 samples show porous hair-like morphology for the Co3O4 shell. The measured shell thickness values are 37 ± 3 nm, 63 ± 8 nm, for S1 and S2, respectively. The estimated void space between the core and the shell lies between 40 and 45 nm and 64 and 72 nm for the SiO2@Co3O4 samples S1 and S2, respectively. The thickness of hair-like structure of the Co3O4 shell was found as 27 ± 3 nm and 45 ± 5 nm for the samples S1 and S2, respectively. The SAED patterns of pure Co3O4 and SiO2@Co3O4 core–shell nanorattles (S1 & S2) are shown in Fig. 6b. The SAED patterns show spots/rings indicating polycrystalline nature of the samples. The SAED patterns could be indexed to (111), (220), (311), (400), (511), and (440) planes of cubic Co3O4. Pure Co3O4 exhibits stronger electron diffraction compared to the SiO2@Co3O4 nanorattles (S1 & S2). The less crystalline nature of SiO2@Co3O4 samples compared to pure Co3O4 suggest that the Co3O4 (shell) particles formed on the silica are smaller in size.
 | ||
| Fig. 6 (a) TEM images of (i) SiO2, (ii) Co3O4, and (iii and iv) SiO2@Co3O4 samples (S1, and S2). The images are obtained for all the samples after calcination at 500 °C. Scale bar is 200 nm. (b) SAED patterns of (i) Co3O4, and (ii and iii) SiO2@Co3O4 samples (S1, and S2). | ||
The specific surface area of pure SiO2, Co3O4 and the SiO2@Co3O4 nanorattles (S1 & S2) were measured using Brunauer–Emmett–Teller (BET) measurements using nitrogen gas physisorption. Pure SiO2 and Co3O4 possess surface area of 34.2 and 46.5 m2 g−1, respectively. The SiO2@Co3O4 nanorattles (S1 and S2) possess surface area of about 279 m2 g−1 and 268 m2 g−1, respectively. The higher surface area of SiO2@Co3O4 nanorattles (S1 and S2) compared to pure Co3O4 and SiO2 is attributed to the nanorattle morphology of SiO2@Co3O4 (S1 and S2). The nanorattles consist of porous shell along with the interior void space between the core and the shell.
The optical properties of pure Co3O4 and SiO2@Co3O4 nanorattles (S1 & S2) were studied using UV-Vis diffuse reflectance spectroscopy (Fig. 7a and b). All the samples exhibit two broad bands in the region 420–450 nm and 700–750 nm. Co3O4 shows absorption bands at 745 nm and 450 nm while SiO2@Co3O4 samples (S1 & S2) show absorption bands at about 705 nm and 425 nm. The band at 745 nm in pure Co3O4 and 705 nm in SiO2@Co3O4 samples (S1 & S2) are due to O2− → Co3+ charge transfer transition and the band at 450 nm in pure Co3O4 and 425 nm in SiO2@Co3O4 samples (S1 & S2) are due to O2− → Co2+ charge transfer transition.19,57 Both the SiO2@Co3O4 samples (S1 & S2) show blue shift of about 40 nm with respect to the 745 nm band in pure Co3O4 and 25 nm with respect to the 450 nm band in pure Co3O4. The blue shift in the case of SiO2@Co3O4 samples (S1 & S2) is attributed to quantum confinement effect and smaller particle size of Co3O4.19,57 Co3O4 is a p-type semiconductor and the exact band gap energies are calculated using the Tauc equation which can be expressed as αhν = k (hν − Eg)n, where Eg represents the band gap, hν is the photon energy, k is the constant, α is the Kubelka–Munk function and n is dependent on the type of transition involved. In the present case, n = 1/2 gives the best fit for (αhν)1/n versus photon energy plots suggesting direct allowed transition in Co3O4. The calculated band gap energy (Eg) values for Co3O4 is 1.70 eV and 2.43 eV and for the SiO2@Co3O4 samples, the values were found to be 1.72 eV and 2.52 eV (S1) and 1.74 eV and 2.57 eV (S2). The band gap values for bulk Co3O4 are 1.48 eV and 2.19 eV.58 The obtained band gap values for SiO2@Co3O4 samples (S1 & S2) are in good agreement with the previously reported values for Co3O4 nanoparticles.19,59
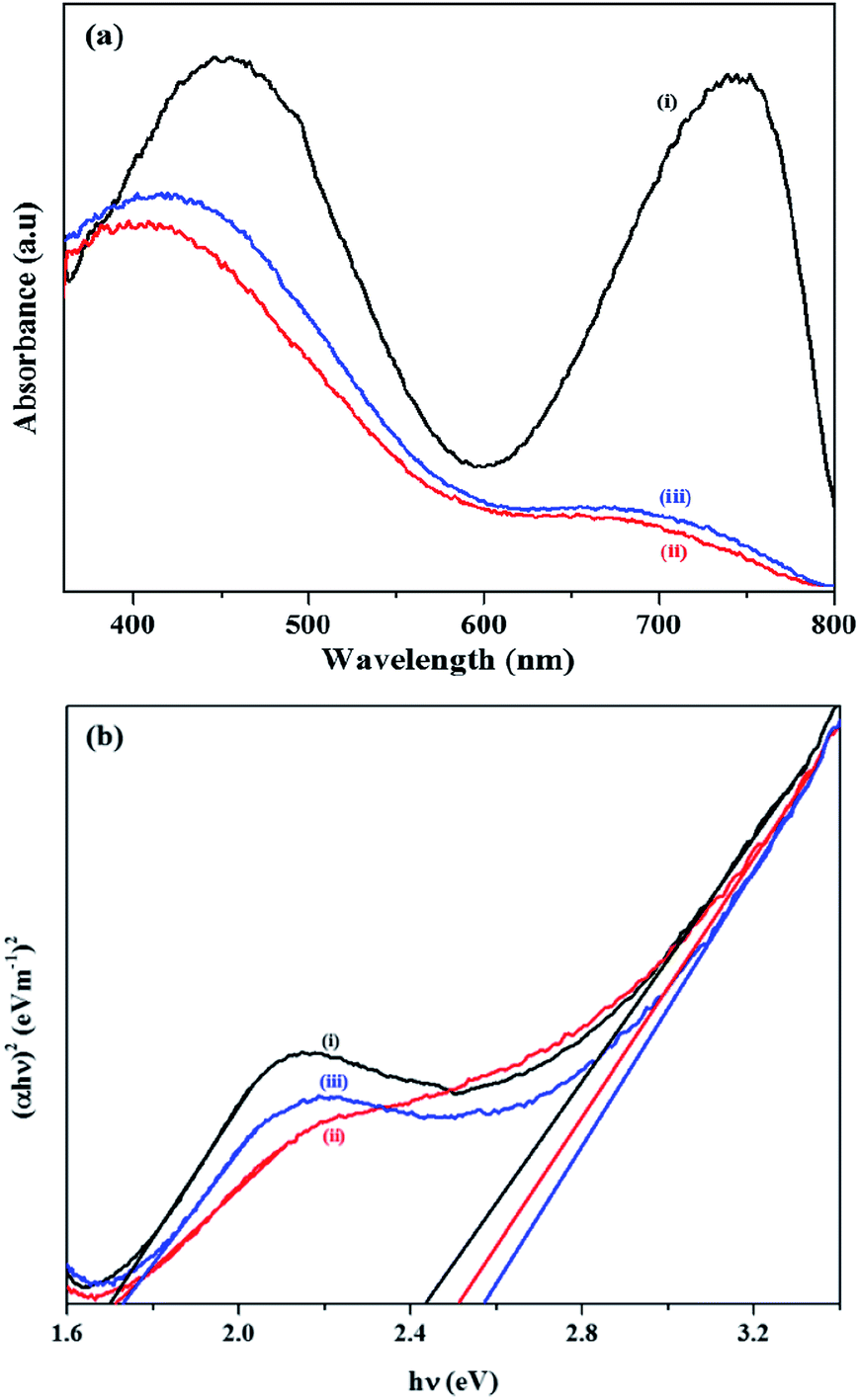 | ||
| Fig. 7 (a) UV-Vis DRS spectra of (i) Co3O4, and (ii and iii) SiO2@Co3O4 core–shell nanorattles (S1 and S2). (b) Tauc plots of (i) Co3O4, and (ii and iii) SiO2@Co3O4 core–shell nanorattles (S1 and S2). | ||
3.2 Proposed mechanism for the formation of SiO2@Co3O4 core–shell nanorattles
For the formation of Co3O4 shell on the SiO2 core, the following mechanism is proposed based on the experimental results (Scheme 1). It is proposed that an adsorption–nucleation–coalescence–anisotropic growth-self-assembly occurs.60,61 At first, Co2+ ions from the solution are adsorbed on the surface of SiO2 through electrostatic interactions. Urea decomposes to produce NH3 and CO2 and the released NH3 readily reacts with the available Co2+ ions on the surface of SiO2 to form a complex (i.e. Co(NH3)y; y ≤ 2) which reduces the concentration of free Co2+ ions in the solution. After attaining the optimum temperature (e.g. 85 °C), OH− ions are steadily produced through the hydrolysis of urea, which is favorable for nucleation and formation of α-Co(OH)2 on the surface of SiO2 spheres based on the coalescence mechanism. The α-Co(OH)2 further undergoes Ostwald ripening to produce flower-like hierarchical α-Co(OH)2 on the surface of SiO2.61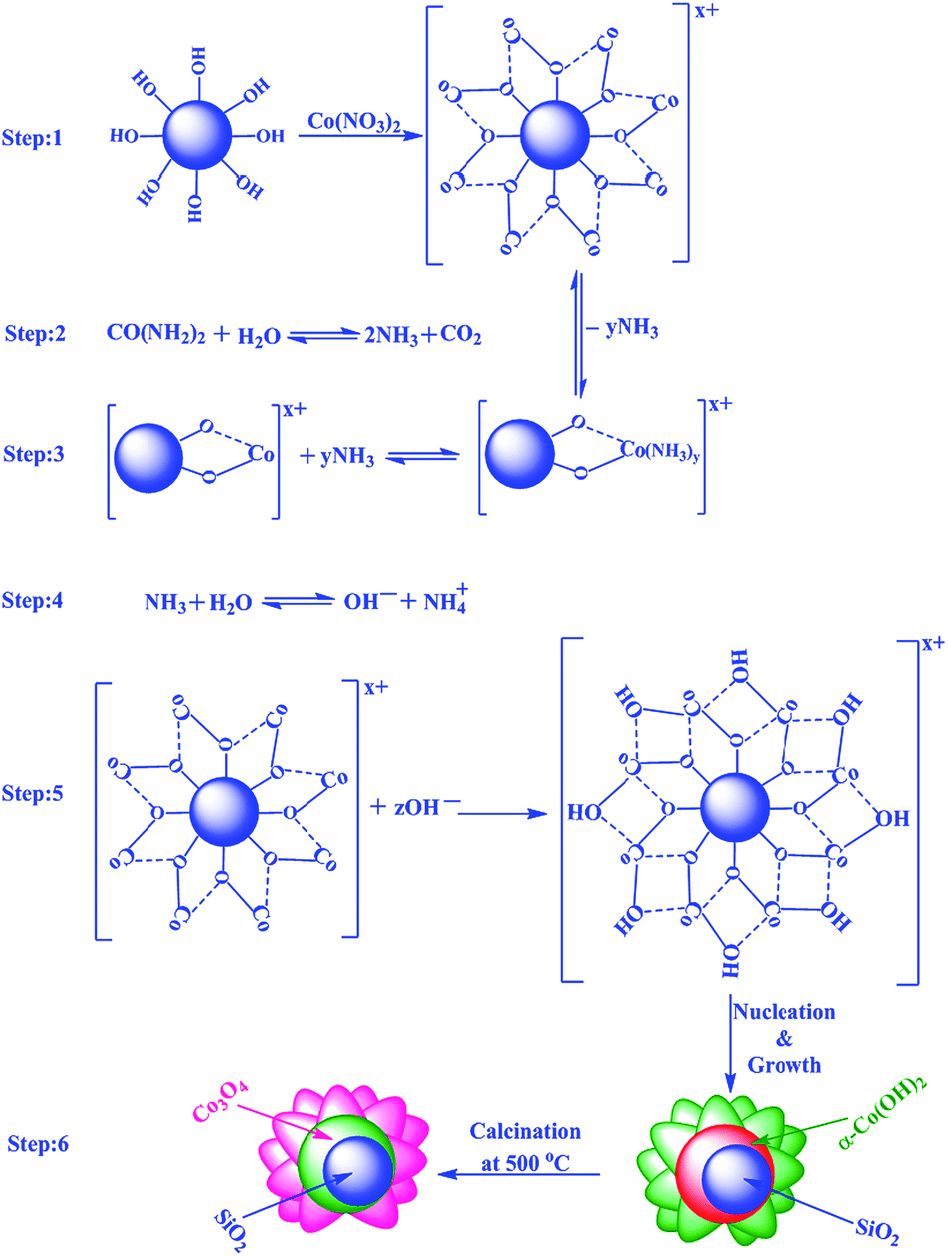 | ||
| Scheme 1 Proposed mechanism for the formation of SiO2@Co3O4 core–shell nanorattles. | ||
Yan et al. and Yuan et al. have explained the formation of hierarchical 3D metal hydroxide flower-like structures based on thermodynamic perspective.60,61 If one considers a single nanoplatelet of α-Co(OH)2, the surface energy is very high. In order to reduce the overall surface energy, the formed ultrathin α-Co(OH)2 nanoplatelets tend to aggregate which decreases the surface energy by reducing the unsatisfied bonds at the exposed areas. The formed thinner plates self-assemble spontaneously based on the coalescence mechanism to produce thicker plates. The pH would decrease and Ostwald ripening growth would dominate leading to flower-like hierarchical α-Co(OH)2 on the surface of SiO2. In the final step, calcination of SiO2@α-Co(OH)2 in air at 500 °C for 3 h leads to the formation of SiO2@Co3O4 core–shell nanorattles through self-template route retaining its 3D hierarchical flower-like morphology for Co3O4 nanoparticles.
To understand the mechanism of formation of nanorattle structures further, time dependent TEM analysis (Fig. 8) was performed at different time intervals for SiO2@α-Co(OH)2 (S1) (e.g. 2 h, 4 h and 6 h) and SiO2@Co3O4 (e.g. 1 h and 3 h). Initially (e.g. 2 h), α-Co(OH)2 nanoparticles are formed around the SiO2 spheres (Fig. 8a). The crystallites of α-Co(OH)2 present on SiO2 are loosely packed and the crystallites located at the outermost surface act as the nucleation seeds for the recrystallization process. As the reaction proceeds from 2 h to 4 h, inside-out Ostwald ripening process dominates which starts the hollowing process by dissolving the smaller crystallites of α-Co(OH)2 present in the interior region. At 6 h, the outer α-Co(OH)2 crystallites become larger which creates an interstitial space with the formation of a compact shell. Calcination of SiO2@α-Co(OH)2 at 500 °C leads to the formation of SiO2@Co3O4 core–shell nanorattles with the retention of core–shell nanorattles morphology (Fig. 8b). The TEM images show the shrinkage of silica core as well as the Co3O4 shell.
 | ||
| Fig. 8 (a) TEM images of SiO2@α-Co(OH)2 samples synthesized at 2 h, 4 h and 6 h and (b) the images of SiO2@Co3O4 obtained on calcination of SiO2@α-Co(OH)2 at 500 °C for 1 h and 3 h. The scale bar is 200 nm. | ||
The size of the inner core (SiO2) and shell (Co3O4) particles exhibits a rather larger polydispersity compared to pure SiO2 and Co3O4 particles. This is explained as follows. It can be noted from TEM studies that the formation of an interface between SiO2 core and α-Co(OH)2 shell occurs within 2 h. After 4 h, formation of core–shell nanorattles with some extra particles of α-Co(OH)2 occurs and core–shell nanorattles with compact α-Co(OH)2 shell are obtained after 6 h. When SiO2@α-Co(OH)2 core–shell nanorattles are calcined to get the SiO2@Co3O4 core–shell nanorattles, polydispersion of silica core particles with slight shrinkage occurs. Intercalated water molecules and anions (NCO− and NO3−) present in α-Co(OH)2 and surface hydroxyl groups from silica are lost during the conversion of SiO2@α-Co(OH)2 to SiO2@Co3O4 core–shell nanorattles. The polydispersity of SiO2 spheres and Co3O4 shell is attributed to the different dehydroxylation rate of loss of surface hydroxyl groups and other species from silica (core) and α-Co(OH)2 (shell).
3.3 The peroxidase-like redox catalytic activity of SiO2@Co3O4 core–shell nanorattles
Enzymes are made up of proteins and they are the active species in bio-catalysis.7,8 They possess high specificity but they can easily loose their stability in harsh environmental conditions such as high temperature, abnormal pH and in the presence of inhibitors. The lack of stability of natural enzymes provides a chance to explore artificial enzymes in bio-applications. At present, artificial enzymes are very attractive towards bio-catalysis because of their stability and high efficiency under a wide range of temperatures and acidic conditions.10,34,62Horseradish peroxidase (HRP), a natural enzyme, catalyzes the oxidation of substrates (e.g. amines and phenols) in the presence of H2O2.34 Mu et al. and Gao et al. have utilized Co3O4 (ref. 10) and Fe3O4 nanoparticles35 as artificial enzymes for testing the intrinsic peroxidase-like activity towards TMB. Inspired from these reports, peroxidase-like redox activity was examined using the SiO2@Co3O4 core–shell nanorattles (S1, S2 and S3). TMB is a colorless peroxidase substrate which is oxidized by H2O2 very slowly to form a blue coloured product. In a typical experiment, the oxidation of TMB was carried out using Co3O4 nanoparticles and SiO2@Co3O4 core–shell nanorattles (S1, S2 and S3) as the catalysts in the presence of H2O2. The blue colored product formed as a result of the catalytic reaction was monitored by UV-Visible spectroscopy (λmax = 652 nm). A blank reaction was carried out with TMB and H2O2 in the absence of any catalyst. It was found that there is no peroxidase activity in the absence of the catalyst which confirms the role of catalyst in the peroxidase-like activity towards TMB. Although, pure Co3O4 nanoparticles and SiO2@Co3O4 core–shell nanorattles (S1, S2 and S3) produce blue coloured product on reaction with TMB in the presence of H2O2, the blue colour was more intense in the case of SiO2@Co3O4 core–shell nanorattles (S1, S2 and S3) compared to pure Co3O4.
The time dependent catalytic activity for Co3O4 and SiO2@Co3O4 core–shell nanorattles towards TMB oxidation, as indicated by UV-Visible spectral results are shown in Fig. 9a. Their relative catalytic efficiency is shown in Fig. 9b which follows the order: S2 > S3 > S1 > Co3O4 > SiO2. The activity of sample S3 is lower compared to S2 but higher than that of S1 (Fig. 9a).
 | ||
| Fig. 9 (a) Time dependent UV-Visible spectral results indicating the peroxidase-like activity on TMB using SiO2, Co3O4, and SiO2@Co3O4 core–shell nanorattles (S1, S2 and S3) as the catalyst (λmax = 652 nm). (b) Comparison of peroxidase activity of SiO2@Co3O4 core–shell nanorattles (S1, S2 and S3) with that of SiO2 and Co3O4 nanoparticles. | ||
3.4 Effect of physicochemical conditions on the peroxidase-like activity
Any catalytic reaction demands optimum conditions for maximum activity. In the present study, TMB and H2O2 were chosen as the reagents. Effort was made to understand the catalytic activity of SiO2@Co3O4 core–shell nanorattles on various conditions such as amount of the catalyst, temperature, and pH. The amount of catalyst (SiO2@Co3O4, S2) was varied from 5 μg to 60 μg per 3 mL while keeping the pH of acetate buffer at 5, temperature at 30 °C and the concentration of H2O2 at 100 mM. A linear relationship between the amount of catalyst and peroxidase activity was found (Fig. 10a). The catalytic activity was tested by varying the pH from 2 to 12 and also by varying the temperature between 20 and 70 °C while keeping the amount of catalyst (SiO2@Co3O4, S2) at 30 μg per 3 mL, and the concentration of TMB and H2O2 at 0.3 mM and 100 mM, respectively. From the results (Fig. 10b and c), the optimized pH and temperature were found as 5 and 30 °C, respectively. The effect of H2O2 concentration on the catalytic activity of SiO2@Co3O4 (S2) was also studied. By increasing the H2O2 concentration, the peroxidase-like activity of the SiO2@Co3O4 core–shell nanorattles is steady even at higher H2O2 concentrations (Fig. 10d). Previous reports have indicated that higher concentration of H2O2 exhibits negative impact on the activity. At higher concentrations, H2O2 inhibits the catalytic activity of HRP due to its conversion from active form to an inactive form.10,63 Leaching test was also performed to prove that the peroxidase-like activity of the catalysts does not result from the leaching of cobalt ions from the catalysts into the solution. To prove this, 30 μg of SiO2@Co3O4 (S2) was incubated in 3 mL of acetate buffer solution (pH = 5) for 30 minutes, and then the SiO2@Co3O4 sample was separated from the buffer solution by centrifugation. The catalytic activity using the resultant solution (from the leaching) was tested (3 mL of solution, 300 μL of 3 mM TMB, and 31 μL of 100 mM H2O2). The peroxidase activity using the leached solution was compared with that of SiO2@Co3O4 sample (S2). Negligible absorbance was found as shown in Fig. 10e when the leached solution was used as the catalyst. These results conclude that the peroxidase-like activity is only due to Co3O4 nanoparticles and not from the leached cobalt ions.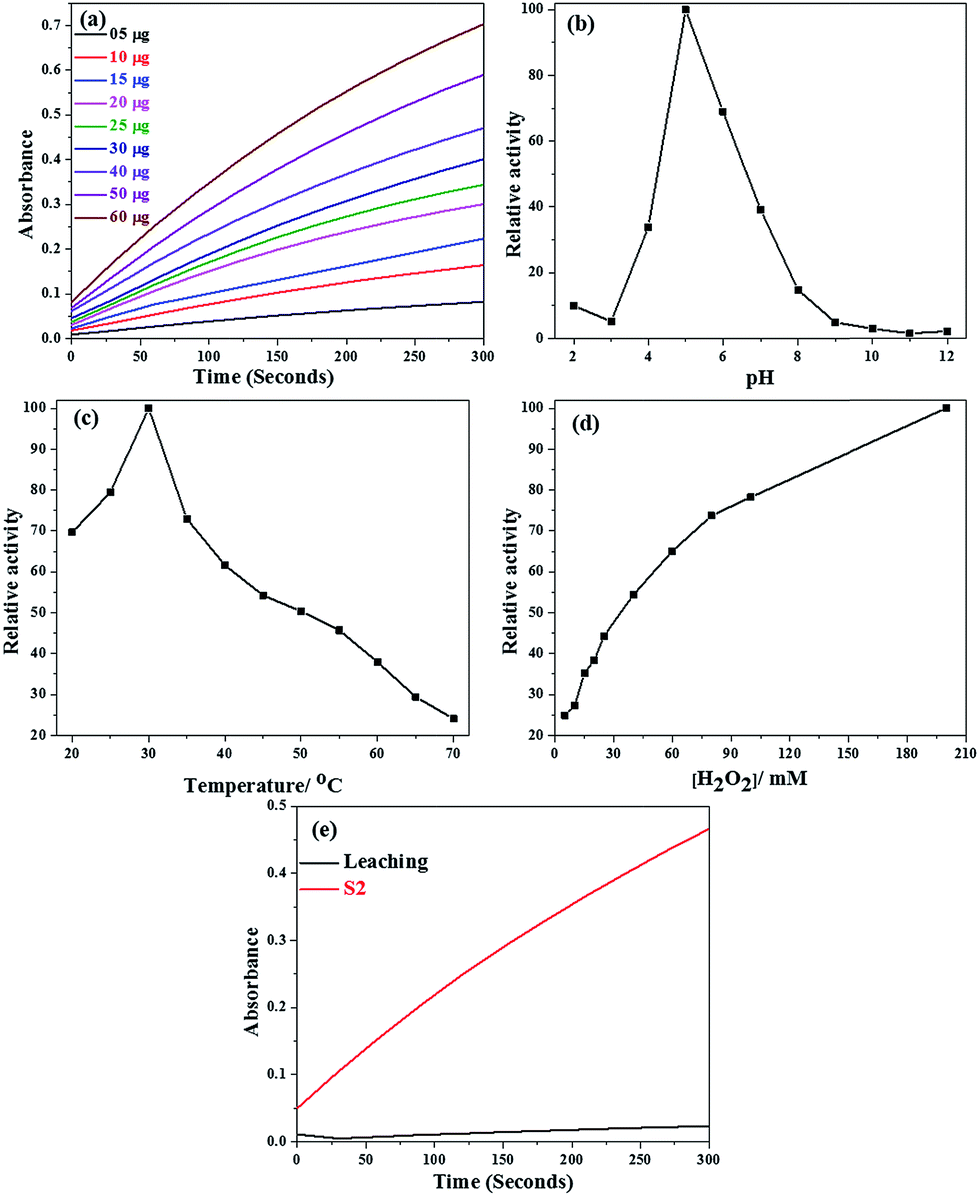 | ||
| Fig. 10 The effect of physicochemical conditions on peroxidase-like activity of SiO2@Co3O4 core–shell nanorattles (S2): (a) amount of catalyst, (b) pH, (c) temperature, (d) H2O2 concentration, and (e) the effect of leaching. | ||
3.5 Steady-state kinetic studies using SiO2@Co3O4 core–shell nanorattles
To understand the peroxidase-like activity of SiO2@Co3O4 core–shell nanorattles better, steady state kinetic analysis was carried out by choosing TMB and H2O2 as the reactants. The kinetic analysis was carried out by varying the concentration of TMB while keeping the H2O2 concentration constant, and vice versa (Fig. 11). The steady state kinetic parameters (Michaelis constant, Km and maximal reaction velocity, Vmax) were estimated using the Lineweaver–Burk double reciprocal plots9,42 which show linear relationship between 1/[V] and 1/[S], as shown in Fig. 11c and d. The estimated kinetic parameters for the SiO2@Co3O4 core–shell nanorattles (S2) were compared with that of Co3O4 nanoparticles and HRP (Table 2). | ||
| Fig. 11 Steady state kinetic analysis for SiO2@Co3O4 core–shell nanorattles (S2) under various conditions: (a) [TMB] = 0.3 mM; [H2O2] = 20–200 mM, (b) [H2O2] = 100 mM; [TMB] = 0.015–0.4 mM, and (c & d) are the Lineweaver–Burk double reciprocal plots corresponding to conditions (a) and (b). Other conditions: 30 μg catalyst per 3 mL of 0.1 M acetate buffer, pH = 5, and temperature = 30 °C. | ||
| Catalyst | [E]/Ma | Substrate | Km/mM | Vmax/Ms−1 (10−8) | kcat/s−1 | kcat/Km (M−1 s−1) | Reference |
|---|---|---|---|---|---|---|---|
| a [E] = the concentration of catalyst (e.g. SiO2@Co3O4) or the concentration of enzyme (HRP). | |||||||
| SiO2@Co3O4 (S2) | 7.54 × 10−11 | TMB | 0.087 ± 0.0028 | 0.012 ± 0.00056 | 1.49 × 104 | 1.75 × 108 | Present work |
| SiO2@Co3O4 (S2) | 7.54 × 10−11 | H2O2 | 25.2 ± 1.28 | 0.015 ± 0.00035 | 1.96 × 104 | 8.08 × 105 | Present work |
| Co3O4 | 3.43 × 10−10 | TMB | 0.037 | 6.27 | 1.83 × 102 | 4.94 × 106 | 10 |
| Co3O4 | 3.43 × 10−10 | H2O2 | 140.07 | 12.1 | 3.53 × 102 | 2.52 × 103 | 10 |
| HRP | 2.50 × 10−11 | TMB | 0.434 | 10.0 | 4.00 × 103 | 9.22 × 106 | 62 |
| HRP | 2.50 × 10−11 | H2O2 | 3.70 | 8.71 | 3.48 × 103 | 9.40 × 105 | 62 |
The Michaelis constant (Km), obtained from the slope of Lineweaver–Burk double reciprocal plot, is related to properties of the substrate, enzyme and the reaction conditions.9 A smaller Km value indicates higher affinity between the substrate and the enzyme. The Km value for SiO2@Co3O4 core–shell nanorattles (S2) is 0.087 for TMB and 25.2 for H2O2, respectively. The Km values for TMB and H2O2 are comparable with that of Co3O4–carbon nitride nanotubes42 (0.056 for TMB and 30.04 for H2O2) and Co3O4/reduced graphene oxide (0.19 for TMB and 24.04 for H2O2).43 The obtained results were compared with the literature values for pure Co3O4 nanoparticles and HRP.10,62 The SiO2@Co3O4 core–shell nanorattles (S2) exhibit five times higher affinity for TMB than HRP and almost two times lower affinity for TMB compared to pure Co3O4 nanoparticles. In the case of H2O2, the SiO2@Co3O4 core–shell nanorattles (S2) show almost six times higher affinity compared to Co3O4 nanoparticles and six times lower affinity compared to HRP. Higher concentration of H2O2 and lower concentration of TMB are the best conditions to achieve maximum peroxidase-like activity using the SiO2@Co3O4 core–shell nanorattles. The higher activity of the nanorattles compared to HRP is attributed to the availability of more active sites on the surface of SiO2@Co3O4 core–shell nanorattles compared to HRP, which has only one active site iron(II).35,62
In order to compare the peroxidase-like activity of SiO2@Co3O4 core–shell nanorattles (S2) with that of natural peroxidase (HRP) and pure Co3O4 nanoparticles, kcat values were also estimated (Table 2). The kcat value for SiO2@Co3O4 core–shell nanorattles (S2) is much higher than that of pure Co3O4 and HRP. These results suggest that the SiO2@Co3O4 core–shell nanorattles (S2) can be used as highly efficient peroxidase mimic compared to pure Co3O4 nanoparticles and HRP.10,62 The ratio of kcat/Km is often referred to as specificity constant; a higher specificity constant value indicates higher peroxidase-like activity of the material. The kcat/Km values estimated for SiO2@Co3O4 core–shell nanorattles (S2) are given in Table 2. In the present study, the SiO2@Co3O4 core–shell nanorattles show higher specificity constant compared to pure Co3O4 and HRP for TMB. With respect to H2O2, the SiO2@Co3O4 core–shell nanorattles (S2) show two orders of magnitude higher specificity compared to pure Co3O4 nanoparticles and the specificity is similar to that of HRP.62 These results demonstrate that the SiO2@Co3O4 core–shell nanorattles show higher peroxidase-like activity compared to Co3O4 nanoparticles and HRP.
3.6 Proposed mechanism for the peroxidase-like activity on TMB using SiO2@Co3O4 core–shell nanorattles
The proposed mechanism for the peroxidase-like activity of SiO2@Co3O4 core–shell nanorattles on TMB is given in Scheme 2.10,42,43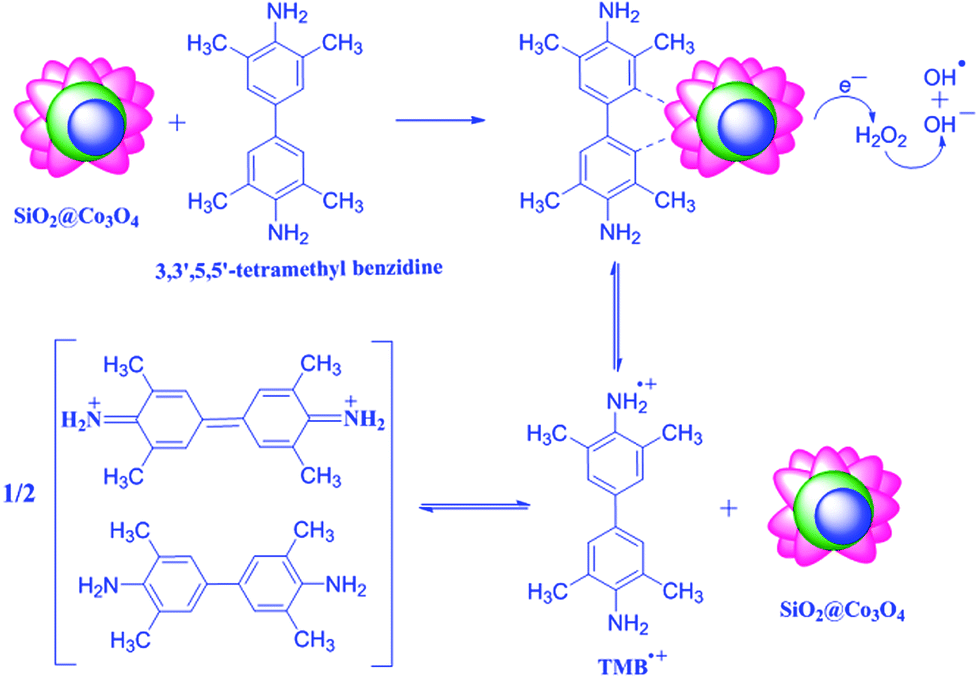 | ||
| Scheme 2 Proposed mechanism for the peroxidase-like activity on TMB using SiO2@Co3O4 core–shell nanorattles. | ||
Neither H2O2 nor SiO2@Co3O4 core–shell nanorattles alone was found to exhibit the peroxidase-like activity. The presence of SiO2@Co3O4 and H2O2 together are important for the peroxidase-like activity on tetramethyl benzidine. When SiO2@Co3O4, H2O2 and TMB are present, the TMB molecules adsorb on the surface of SiO2@Co3O4 and increases the electron density by donating the lone pair electrons of the amino groups.10,64 XPS or WAXS measurements would give more evidence on this aspect and this would be part of future studies. The SiO2@Co3O4 core–shell nanorattles induce electrochemical reduction of H2O2 producing OH− and OH˙ species which involve in the oxidation of TMB to produce TMB˙+ (a charge transfer complex which is blue in color). Mu et al. have reported the electrochemical reduction of H2O2 using pure Co3O4 nanoparticles.10 In the case of core–shell nanorattles, there is an assembly of Co3O4 nanoparticles on the surface of SiO2 spheres. The TMB molecules undergo less steric hindrance to reach the catalytic active centers (cobalt) in the case of SiO2@Co3O4 core–shell nanorattles compared to pure Co3O4 nanoparticles. This increases the electron density on the core–shell nanorattles and facilitate the electron transfer. The enhancement in the peroxidase-like activity of SiO2@Co3O4 core–shell nanorattles compared to pure Co3O4 nanoparticles is due to increased surface area and also due to increase in the electron density in the case of SiO2@Co3O4 core–shell nanorattles compared to pure Co3O4 nanoparticles. Sample S2 shows higher activity than S1 and this is due to the presence of more number of catalytic active centers in S2 compared to S1 but sample S3 consists of free Co3O4 nanoparticles in addition to SiO2@Co3O4 nanorattles which leads to lower activity compared to S2.
4. Conclusions
A novel self-template method for the synthesis of SiO2@Co3O4 core–shell nanorattles with different shell thickness has been reported. Compared to other preparation methods, this approach does not require any surface modification and the method is facile, and inexpensive. The synthesized SiO2@Co3O4 core–shell nanorattles were characterized using a variety of techniques. The SiO2@Co3O4 core–shell nanorattles possess enhanced intrinsic peroxidase-like activity compared to pure Co3O4 nanoparticles and HRP and it follows Michaelis–Menten kinetics. The enhanced peroxidase-like activity of SiO2@Co3O4 core–shell nanorattles is due to the synergistic interaction between the SiO2 core and the Co3O4 shell. The nanorattles show higher kcat and kcat/Km values compared to pure Co3O4 nanoparticles and HRP suggesting their applicability as an artificial enzyme mimic in biomedicine and bio-sensing applications.Acknowledgements
Syam Kandula thanks the Ministry of Human Resources and Development (MHRD), Govt. of India for the financial support. The authors are thankful to the Institute Instrumentation Centre, IIT Roorkee for providing the facilities.Notes and references
- X. Xu, B. Tian, J. Kong, S. Zhang, B. Liu and D. Zhao, Adv. Mater., 2003, 15, 1932–1936 CrossRef CAS.
- W. Shi, X. Zhang, S. He and Y. Huang, Chem. Commun., 2011, 47, 10785–10787 RSC.
- W. Chen, J. Chen, Y. B. Feng, L. Hong, Q. Y. Chen, L. F. Wu, X. H. Lin and X. H. Xia, Analyst, 2012, 137, 1706–1712 RSC.
- L. Zhang, L. Han, P. Hu, L. Wang and S. Dong, Chem. Commun., 2013, 49, 10480–10482 RSC.
- C. Ray, S. Dutta, S. Sarkar, R. Sahoo, A. Roy and T. Pal, J. Mater. Chem. B, 2014, 2, 6097–6105 RSC.
- G. Nie, L. Zhang, J. Lei, L. Yang, Z. Zhang, X. Lu and C. Wang, J. Mater. Chem. A, 2014, 2, 2910–2914 CAS.
- W. Zhang, X. Liu, D. Walsh, S. Yao, Y. Kou and D. Ma, Small, 2012, 8, 2948–2953 CrossRef CAS PubMed.
- J. Dong, L. Song, J. J. Yin, W. He, Y. Wu, N. Gu and Y. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 1959–1970 CAS.
- H. Sun, X. Jiao, Y. Han, Z. Jiang and D. Chen, Eur. J. Inorg. Chem., 2013,(1), 109–114 CrossRef CAS.
- J. Mu, Y. Wang, M. Zhao and L. Zhang, Chem. Commun., 2012, 48, 2540–2542 RSC.
- B. J. Jankiewicz, D. Jamiola, J. Choma and M. Jaroniec, Adv. Colloid Interface Sci., 2012, 170, 28–47 CrossRef CAS PubMed.
- Y. Piao, A. Burns, J. Kim, U. Wiesner and T. Hyeon, Adv. Funct. Mater., 2008, 18, 3745–3758 CrossRef CAS.
- C. G. Schaefer, S. Vowinkel, G. P. Hellmann, T. Herdt, C. Contiu, J. J. Schneider and M. Gallei, J. Mater. Chem. C, 2014, 2, 7960–7975 RSC.
- L. Ren, P. Wang, Y. Han, C. Hu and B. Wei, Chem. Phys. Lett., 2009, 476, 78–83 CrossRef CAS PubMed.
- Y. Kim, J. H. Lee, S. Cho, Y. Kwon, I. In, J. Lee, N. H. You, E. Reichmanis, H. Ko, K. T. Lee, H. K. Kwon, D. H. Ko, H. Yang and B. Park, ACS Nano, 2014, 8, 6701–6712 CrossRef CAS PubMed.
- C. Sun, S. Rajasekhara, Y. Chen and J. B. Goodenough, Chem. Commun., 2011, 47, 12852–12854 RSC.
- X. Wang, X. Chen, L. Gao, H. Zheng, Z. Zhang and Y. Qian, J. Phys. Chem. B, 2004, 108, 16401–16404 CrossRef CAS.
- E. Barrera, I. Gonzalez and T. Viveros, Sol. Energy Mater. Sol. Cells, 1998, 51, 69–82 CrossRef CAS.
- K. Deori, S. K. Ujjain, R. K. Sharma and S. Deka, ACS Appl. Mater. Interfaces, 2013, 5, 10665–10672 CAS.
- J. D. Blakemore, H. B. Gray, J. R. Winkler and A. M. Muller, ACS Catal., 2013, 3, 2497–2500 CrossRef CAS.
- N. Kang, J. H. Park, M. Jin, N. Park, S. M. Lee, H. J. Kim, J. M. Kim and S. U. Son, J. Am. Chem. Soc., 2013, 135, 19115–19118 CrossRef CAS PubMed.
- N. Yan, Q. Chen, F. Wang, Y. Wang, H. Zhong and L. Hu, J. Mater. Chem. A, 2013, 1, 637–643 CAS.
- L. Tan, X. Wu, D. Chen, H. Liu, X. Meng and F. Tang, J. Mater. Chem. A, 2013, 1, 10382–10388 CAS.
- P. Yang, F. Wang, X. Luo, Y. Zhang, J. Guo, W. Shi and C. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 12581–12587 CAS.
- B. Zhang, T. Cai, S. Li, X. Zhang, Y. Chen, K. G. Neoh, E. T. Kang and C. Wang, J. Mater. Chem. C, 2014, 2, 5189–5197 RSC.
- X. Liang, J. Li, J. B. Joo, A. Gutierrez, A. Tillekaratne, I. Lee, Y. Yin and F. Zaera, Angew. Chem., Int. Ed., 2012, 51, 8034–8036 CrossRef CAS PubMed.
- P. H. C. Camargo, Z. Y. Li and Y. Xia, Soft Matter, 2007, 3, 1215–1222 RSC.
- D. Scheid, G. Cherkashinin, E. Ionescu and M. Gallei, Langmuir, 2014, 30, 1204–1209 CrossRef CAS PubMed.
- J. Xu, J. Liu, R. Che, C. Liang, M. Cao, Y. Li and Z. Liu, Nanoscale, 2014, 6, 5782–5790 RSC.
- W. Na, Q. Wei and Z. Nie, J. Mater. Chem., 2012, 22, 9970–9974 RSC.
- A. Agiral, H. S. Soo and H. Frei, Chem. Mater., 2013, 25, 2264–2273 CrossRef CAS.
- R. Xie, C. Wang, L. Xia, H. Wang, T. Zhao and Y. Sun, Catal. Lett., 2014, 144, 516–523 CrossRef CAS.
- J. Zong, Y. Zhu, X. Yang and C. Li, J. Alloys Compd., 2011, 509, 2970–2975 CrossRef CAS PubMed.
- J. Mu, L. Zhang, G. Zhao and Y. Wang, Phys. Chem. Chem. Phys., 2014, 16, 15709–15716 RSC.
- L. Gao, J. Zhuang, L. Nie, J. Zhang, Y. Zhang, N. Gu, T. Wang, J. Feng, D. Yang, S. Perrett and X. Yan, Nat. Nanotechnol., 2007, 2, 577–583 CrossRef CAS PubMed.
- H. Chen, Y. Li, F. Zhang, G. Zhang and X. Fan, J. Mater. Chem., 2011, 21, 17658–17661 RSC.
- X. Chen, X. Tian, B. Su, Z. Huang, X. Chen and M. Oyama, Dalton Trans., 2014, 43, 7449–7454 RSC.
- L. Chen, B. Sun, X. Wang, F. Qiao and S. Ai, J. Mater. Chem. B, 2013, 1, 2268–2274 RSC.
- K. Cai, Z. Lv, K. Chen, L. Huang, J. Wang, F. Shao, Y. Wang and H. Han, Chem. Commun., 2013, 49, 6024–6026 RSC.
- G. Nie, L. Zhang, X. Lu, X. Bian, W. Sun and C. Wang, Dalton Trans., 2013, 42, 14006–14013 RSC.
- H. Wang, S. Li, Y. Si, Z. Sun, S. Li and Y. Lin, J. Mater. Chem. B, 2014, 2, 4442–4448 RSC.
- C. O. Song, J. W. Lee, H. S. Choi and J. K. Jeung, RSC. Adv., 2013, 3, 20179–20185 RSC.
- J. Xie, H. Cao, H. Jiang, Y. Chen, W. Shi, H. Zheng and Y. Huang, Anal. Chim. Acta, 2013, 796, 92–100 CrossRef CAS PubMed.
- S. Kandula and P. Jeevanandam, J. Alloys Compd., 2014, 615, 167–176 CrossRef CAS PubMed.
- Z. Liu, R. Ma, M. Osada, K. Takada and T. Sasaki, J. Am. Chem. Soc., 2005, 127, 13869–13874 CrossRef CAS PubMed.
- W. Chen, Y. Yang, H. Shao and J. Fan, J. Phys. Chem. C, 2008, 112, 17471–17477 CAS.
- J. P. Cheng, L. Liu, J. Zhang, F. Liu and X. B. Zhang, J. Electroanal. Chem., 2014, 722–723, 23–31 CrossRef CAS PubMed.
- Q. Wang, S. Liu, H. Sun and Q. Lu, Ind. Eng. Chem. Res., 2014, 53, 7917–7922 CrossRef CAS.
- Y. Oaki, S. Kajiyama, T. Nishimura and T. Kato, J. Mater. Chem., 2008, 18, 4140–4142 RSC.
- P. N. R. Kishore and P. Jeevanandam, J. Nanosci. Nanotechnol., 2013, 13, 2908–2916 CrossRef CAS PubMed.
- R. L. Frost, D. L. Wain, W. N. Martens and B. J. Reddy, Spectrochim. Acta, Part A, 2007, 66, 1068–1074 CrossRef PubMed.
- N. Bayal and P. Jeevanandam, J. Nanopart. Res., 2013, 15, 2066/1–2066/15 CrossRef.
- D. Ghosh, S. Giri and C. K. Das, ACS Sustainable Chem. Eng., 2013, 1, 1135–1142 CrossRef CAS.
- Z. Z. Xu, Z. L. Chen, Y. Ben and J. M. Shen, Mater. Lett., 2009, 63, 1210–1212 CrossRef CAS PubMed.
- S. J. Palmer, R. L. Frost and T. Nguyen, Coord. Chem. Rev., 2009, 253, 250–267 CrossRef CAS PubMed.
- F. Malherbe and J. P. Besse, J. Solid State Chem., 2000, 155, 332–341 CrossRef CAS.
- K. Deori and S. Deka, CrystEngComm, 2013, 15, 8465–8474 RSC.
- S. Farhadi, K. Pourzare and S. Bazgir, J. Alloys Compd., 2014, 587, 632–637 CrossRef CAS PubMed.
- A. Gulino, P. Dapporto, P. Rossi and I. Fragala, Chem. Mater., 2003, 15, 3748–3752 CrossRef CAS.
- J. Yan, Z. Fan, W. Sun, G. Ning, T. Wei, Q. Zhang, R. Zhang, L. Zhi and F. Wei, Adv. Funct. Mater., 2012, 22, 2632–2641 CrossRef CAS.
- C. Yuan, X. Zhang, L. Su, B. Gao and L. Shen, J. Mater. Chem., 2009, 19, 5772–5777 RSC.
- X. Q. Zhang, S. W. Gong, Y. Zhang, T. Yang, C. Y. Wang and N. Gu, J. Mater. Chem., 2010, 20, 5110–5116 RSC.
- J. A. Nicell and H. Wright, Enzyme Microb. Technol., 1997, 21, 302–310 CrossRef CAS.
- Y. Song, K. Qu, C. Zhao, J. Ren and X. Qu, Adv. Mater., 2010, 22, 2206–2210 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Overview image of SiO2@Co3O4 core–shell nanorattles. See DOI: 10.1039/c4ra12596g |
| This journal is © The Royal Society of Chemistry 2015 |