
Density functional theory calculations of atomic, electronic and thermodynamic properties of cubic LaCoO3 and La1−xSrxCoO3 surfaces†
Abstract
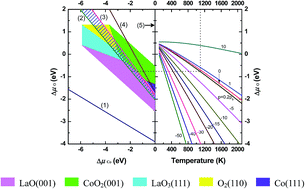
Spin-polarized density functional theory calculations employing the generalized gradient approximation (GGA) + U scheme were used to investigate the atomic, and electronic structures of ferromagnetic LaCoO3 and La1−xSrxCoO3 surfaces. Nonstoichiometric symmetrical slab models were adopted. The thermodynamic stability of these surfaces was analyzed with phase diagrams drawn with the total energies obtained from calculations. The influence of Sr-doping on the thermodynamic stability of the surfaces was also examined. The results indicate that Sr ions prefer to substitute La ions in the outermost layers rather than those in bulk layers for both (001) surfaces. In the undoped LaCoO3 system, CoO2- and LaO-terminated (001) surfaces are the most stable of all the considered surfaces under typical operational conditions of the solid oxide fuel cells (T = 1100 K, pO2 = 0.2 atm), while Sr-doping in the LaCoO3 crystal destabilizes the CoO2-terminated surface with respect to the La0.75Sr0.25O-terminated surface.
 Please wait while we load your content...
Please wait while we load your content...

