
Preparation of large micron-sized monodisperse polystyrene/silver core–shell microspheres with compact shell structure and their electrical conductive and catalytic properties†
Yougen Huab,
Tao Zhaoa,
Pengli Zhu*a,
Xianwen Lianga,
Rong Sun*a and
Ching-Ping Wongcd
aShenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen, China. E-mail: pl.zhu@siat.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, China
cSchool of Materials Sciences and Engineering, Georgia Institute of Technology, Atlanta, USA
dDepartment of Electronic Engineering, The Chinese University of Hong Kong, Hong Kong, China
First published on 1st December 2014
Abstract
Utilizing a simple improved electroless plating method, ca. 6 μm sized monodisperse polystyrene/silver (PS/Ag) core–shell microspheres with a complete, homogeneous and compact coverage of Ag nanoparticles layer were successfully prepared. In this approach, modified lightly cross-linked PS (LCPS) microspheres with a uniform diameter of 5.6 μm were used as templates. After the LCPS cores were removed by dissolving with dimethyl formamide (DMF), the outer silver shells assembled by amounts of Ag nanoparticles maintain good hollow spherical structure. The size and coverage degree of Ag nanoparticles on the PS/Ag microspheres could be easily tuned by changing the concentration of [Ag(NH3)2]+ ions in aqueous media. The electrical conductivity of the obtained PS/Ag core–shell microspheres changes from 3.76 × 104 S m−1 to 3.33 × 105 S m−1 as increasing the coverage density of Ag nanoparticles. Moreover, the catalytic assays indicate that the resultant monodisperse PS/Ag core–shell microspheres show excellent catalytic activity for the reduction of methylene blue (MB) by NaBH4. In particular, the corresponding hollow Ag spheres exhibit more outstanding catalytic activity due to their stable hollow structure and higher specific surface area.
1. Introduction
Core–shell structural polymer–metal particles, in general, have dual properties as both organic and metallic materials, and have found ever-increasing applications in a wide variety of scientific and technological applications, such as microelectronics,1,2 catalysis,3–5 surface-enhanced Raman scattering,6,7 magnetic,8 and biomedical applications.9,10 Another interesting application of these core–shell structures is for the synthesis of hollow metal particles by using the inner polymeric material as the sacrificial core.11,12Among the polymeric cores, polystyrene (PS) spheres present some major advantages, such as their controlled size, shape and monodispersity, various functional groups and easy removal by calcining at high temperature or dissolving with an appropriate solvent. Actually, various noble metal-coated PS core–shell particles, especially silver-coated PS spheres, have been prepared successfully by different strategies, such as electroless plating,13 electrostatic deposition,14 sputtering methods,15 in situ chemical reduction,16,17 and layer-by-layer (LBL) self-assembly.18 However, in most cases, there still exist various problems, such as the production process was environmentally unfriendly, time-consuming and extreme operating conditions. In addition, these approaches usually not able to fabricate hollow metallic spheres via removing the cores, due to the poor and non-uniform metal coverage or the thin metallic shell, which lack enough mechanical strength to maintain the residue hollow structure. Electroless plating is a widely used method to form the metallic coating on the flat polymer surface, but it is still difficult to coat a uniform metal layer onto the surface of polymer spheres because of the existence of hoop stresses.19 Recently, many researchers did a lot of works and try to modify this traditional electroless plating. Wang et al.20 reported a relatively versatile method based on poly(dopamine)-assisted electroless plating to fabricate PS/Ag core–shell composite microspheres. Although the adhesion between the PS and silver was improved, aggregation of PS/Ag microspheres occurred because of the irregularity of silver shell. Ma et al.13 prepared PS/Ag composite microsphere with controllable silver shell thickness via modified electroless plating. But its laborious procedures were tedious and toxic reagents, such as chloroplatinic acid were used. Cai et al.17 fabricated hollow silver spheres using sulfonated polystyrene spheres as template via silver-mirror reaction at low temperature. Although the hollow compact silver spheres were obtained, the silver nanoparticles on the surfaces of PS cores were easily fall off due to the poor adhesion between the core and outer silver nanoparticles.
Moreover, to the best of our knowledge, the successful synthetic core–shell polymer–metal spheres were mostly used polymeric spheres with diameter of submicron or micron-sized within 4 μm as templates,3–7,13,14,21 polymer cores diameter beyond 4 μm were rarely reported.22,23 Large micron-sized spheres have many advantages, for example, easily separable and recyclable by centrifugation, avoidable agglomerations, visible and thus can be easily monitored by optical methods, which are suitable for some special applications, such as serve as spacers for display panel and conductive supporters. Micron-sized monodisperse PS spheres can be synthesized by seeded polymerization, distillation precipitation polymerization and some specific technology with the aid of sophisticated instrument, such as micro-fluidic devices, jet break-up devices and Shirasu Porous Glass (SPG) membrane.24–26 However, these processes are usually complex, time-consuming, costly and environment unfriendly. Dispersion polymerization is a simple and effective polymerization method to prepare micron-sized monodisperse PS spheres. Nevertheless, the preparation of monodisperse PS microspheres, especially cross-linked PS microspheres, with large size beyond 5 μm is still a challenging work via dispersion polymerization method. It is well known that the cross-linking agent, such as DVB, could interfere with the dispersion polymerization of styrene in ethanol, inappropriate amounts of DVB would broaden the particle size distribution and lead to irregular particles or coagulation. Song et al.27 and Cao et al.28 fabricated cross-linked PS spheres with diameters ranging from 2 μm to 4 μm by utilizing two-stage dispersion polymerization method. Qi and co-workers25 prepared large-sized monodisperse PS microspheres in the size range of 3.75–7.09 μm by dispersion polymerization with dropwise monomer feed procedure. In our study, monodisperse LCPS microspheres with an average diameter of 5.6 μm were successfully synthesized by one-step dispersion polymerization, which simplified the process of preparation of large micron-sized monodisperse PS spheres.
Herein, we report a simple, efficient and environmentally friendly method to prepare monodisperse 5 μm sized PS/Ag core–shell microspheres with full and compact coverage of Ag nanoparticles shells. The obtained PS/Ag core–shell hybrid microspheres were investigated by SEM, FTIR, XPS, XRD and TGA. Four-point probe system was employed for characterization of the electrical conductive properties of PS/Ag hybrid microspheres. In addition, the catalytic activity of the produced PS/Ag core–shell microspheres and the corresponding hollow Ag spheres used as catalysts to reduce methylene blue in the presence of NaBH4 were also studied in detail.
2. Experimental
2.1. Materials
Styrene was purchased from Shanghai Chemical Reagent Co. (China) and purified by treating with 5% NaOH aqueous solution to remove the inhibitor. AgNO3 (99.8%), H2SO4 (98%), NH3·H2O (28%), NaBH4, poly(vinyl pyrrolidone) (PVP, K-30), tin(II) chloride dihydrate (SnCl2·2H2O), azobisisobutyronitrile (AIBN), sodium potassium tartrate (C4H4O6KNa·4H2O) and methylene blue (MB, C16H18ClN3S) were supplied by Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China) and used as received. Divinylbenzene (DVB, 50%) was obtained from Tokyo Chemical Industry (TCI, Shanghai). Ultrapure water (>18 MΩ cm−1) was used for all experiments.2.2. Synthesis of monodisperse LCPS microspheres
Monodisperse lightly cross-linked PS (LCPS) microspheres with an average diameter of 5.6 μm were synthesized via one-step dispersion polymerization method. The typical process was carried out as follows: St (20.0 g), DVB (0.1 g) and AIBN (0.6 g) were dissolved in a mixture of PVP (0.6 g), ethanol (47.5 g) and H2O (2.5 g). The above mixture was conducted in a four necked round-bottom flask (150 mL) fitting with mechanical stirrer, thermometer, nitrogen inlet and reflex condenser, and then heated to 70 °C for 24 h with an agitation speed of 200 rpm. The reaction solution was deoxygenated by bubbling nitrogen gas throughout the polymerization. After reaction, the crude suspension was purified by successive centrifugation, decantation and redispersion with ultrapure water and ethanol for at least three times. Then the purified products were dried at room temperature in a vacuum oven for further use.2.3. Preparation of monodisperse PS/Ag core–shell microspheres
First of all, the LCPS microspheres were sulfonated by concentrated sulfuric acid. In detail, 2.5 g as-prepared LCPS powders were immersed in 50 mL concentrated sulfuric acid at 40 °C for 4 h with a constant stirring rate at 100 rpm to obtain negatively charged sulfonated PS (SPS) particles. The suspension was centrifuged and washed with ethanol several times, and then redispersed into 50 mL ultrapure water for subsequent use. Afterward, 50 mL SPS dispersion was mixed with 50 mL acidic SnCl2 aqueous solution (44 mM SnCl2 and 0.2 M HCl), and stirred at room temperature for 1 h to obtain Sn2+-sensitized SPS dispersion. The mixture was centrifuged and rinsed with ultrapure water to remove the unabsorbed Sn2+ ions. Then, 50 mL fresh [Ag(NH3)2]+ complex ions aqueous solution (15 mM, 30 mM and 60 mM) and 50 mL C4H4O6KNa aqueous solution (20 mM) was successively quickly poured into the dispersion mentioned above, magnetically stirred at a speed of 100 rpm at room temperature for 1 h. After reaction, the products were collected and purified by successive centrifugation and ultrasonic washing with ethanol by a bath ultrasonic cleaner (40 kHz, 500 W) for 5 min at least three times. For convenient description, the PS/Ag samples prepared with different [Ag(NH3)2]+ concentration are labeled as PS/Ag-15, PS/Ag-30 and PS/Ag-60, respectively.To confirm that hollow structures existed, PS cores were removed using DMF, in which the core–shell structural PS/Ag hybrid microspheres were kept for 6 h with stirring at room temperature. The hollow Ag microspheres were then separated from the DMF and rinsed with ethanol and then dried under vacuum at 60 °C.
2.4. Catalytic activity of the monodisperse PS/Ag core–shell microspheres and their corresponding hollow Ag spheres
The catalytic activity of the as-synthesized monodisperse PS/Ag core–shell hybrid microspheres and their corresponding hollow Ag spheres was examined by measuring the reduction of dye MB by NaBH4. The reaction unsupported by the catalyst was studied as a reference. In a typical catalytic experiment, catalysts of the PS/Ag microspheres and the hollow Ag spheres were firstly dispersed into ultrapure water to obtain 2 mg mL−1 dispersion. Then, a given amount of dispersion consisting of catalysts was mixed with 10 mL MB aqueous solutions (0.05 mM) and 10 mL freshly prepared NaBH4 aqueous solutions (25 mM) at ambient temperature with stirring. The UV-Vis absorption spectra during the reduction were recorded with a regular time interval.2.5. Characterization
The morphologies of samples were observed by a field emitting scanning electron microscope (FEI Nova Nano SEM 450, USA) and transmission electron microscopy (TEM, FEI Tecnai G2F20S-TWIN). A FTIR spectrometer (Bruker VERTEX 70, German) was employed to characterize chemical information of the products. Surface chemical information of samples were further measured by a multifunctional X-ray photoelectron spectrometer (Perkin-Elmer, PHI 5702, USA) using an Al Kα X-ray excitation source (1486.6 eV) at a constant recording ratio of 40. X-ray diffraction analysis was carried out on a D/MAX-RA X-ray diffractometer (Rigaku, Japan) with Cu Kα radiation (1.54056 Å) in the range 10° to 90° with a scanning rate of 5° per min. Thermogravimetric analysis was performed on a TG instrument (TA SDT Q600, USA) at a heating rate of 10 °C min−1 from room temperature to 800 °C under a nitrogen atmosphere with a steady flow rate of 100 mL min−1. UV-Vis absorption spectra were recorded on the UV-Vis-NIR spectrometer (Shimadzu UV-3600, Japan) with a wavelength range of 400–800 nm. The Brunauer–Emmett–Teller (BET) surface area was measured by nitrogen adsorption (the relative pressure P/P0 range from 0.05 to 0.35) on a specific surface analyser (Micromeritics ASAP 2020M + C, USA). The bulk electrical conductivity of the as-prepared PS/Ag core–shell microspheres was measured with a digital four-point probe system (RTS-8, Guangzhou four-point probe technology, China) at room temperature. To reduce the influence of the contact resistance, PS/Ag microspheres were compressed into slices using a tablet press machine under 2.5 MPa.3. Results and discussion
3.1. Preparation and characterization of monodisperse PS/Ag core–shell microspheres and their corresponding hollow Ag spheres
The detailed preparation procedure of the monodisperse PS/Ag core–shell microspheres and their corresponding hollow Ag spheres are illustrated detail in Scheme 1. In this approach, the LCPS spheres with large micron-size were firstly synthesized via a one-step dispersion polymerization using a small amount DVB (0.5 wt% based on St monomer) as cross-linking agent (Scheme 1a). The dispersion polymerization is an attractive method for producing micron-size polymer microspheres in a single batch process, and usually leads to microspheres with very narrow size distribution. | ||
| Scheme 1 Schematic diagram illustrates the formation of monodisperse PS/Ag core–shell microspheres and their corresponding hollow Ag spheres. | ||
The SEM image of the as-prepared LCPS spheres are shown in Fig. 1a and confirms that the size of the LCPS particles are uniform with an average diameter of 5.6 μm, and the coefficient of variation is only about 2.3% (by averaging 100 particles in the SEM images). Moreover, the magnified SEM images of the typical LCPS microspheres indicate that they have good spherical shapes and their surfaces are very smooth (Fig. 1b). The results show that the as-synthesized large micron-sized LCPS spheres possess excellent monodispersity, and they are ideal templates for preparation of composite microspheres or hollow microspheres on a commercial scale.
 | ||
| Fig. 1 SEM images of LCPS microspheres. | ||
The as-prepared LCPS microspheres were used as templates and modified by concentrated sulfuric acid to obtain SPS microspheres with –SO3H groups on the surfaces (Scheme 1b). The morphology of the SPS microspheres was examined by SEM (Fig. S1†), which clearly indicates that the sulfonation do not have significant influence on the spherical shape and size of the original LCPS microspheres due to the sulfonation reaction usually occurred at the surfaces. The existence of –SO3H groups on the SPS microspheres was confirmed with the FTIR and shown in Fig. 2. Before the sulfonation reaction, peaks located at 3059, 3024, 2922, 2848 cm−1, and 1601, 1493, 1452 cm−1 can be seen clearly in the FTIR spectrum of LCPS (Fig. 2a), which are the well-defined characteristic bands of the styrene unit.29 After modification with concentrated sulfuric acid, new peaks at 1180 and 1042 cm−1 appear, as shown in the magnified inset in Fig. 2. These new peaks can be ascribed to the asymmetric and symmetric vibration of –SO3H groups.30 All the above peaks observed in the spectra suggest that the –SO3H groups are successfully decorated on the LCPS microspheres after sulfonation reaction.
 | ||
| Fig. 2 FT-IR spectra of (a) LCPS microspheres and (b) SPS microspheres. | ||
An XPS was used to analyze the surface chemical composition of the LCPS and SPS microspheres to further confirm the presence of –SO3H groups on the surfaces of SPS spheres. For the original LCPS, the presence of abundant carbon and oxygen on the surface is revealed by two strong peaks of C1s and O1s at 284.6 eV and 531.8 eV, respectively (Fig. 3a). For the SPS microspheres, in addition to the peaks of C1s and O1s, two relatively weak peaks at 232.6 eV and 168.6 eV are appeared, which corresponding to sulfur of S1s and S2p (Fig. 3b). Moreover, the surface O/C mole ratio of SPS microspheres is higher than that of LCPS microspheres. The results clearly indicate that there are groups containing sulfur and oxygen on the surface of SPS microspheres. All the evidence demonstrates that the –SO3H groups have been successfully connected to the surfaces of LCPS microspheres, which is consistent with the results of FTIR in Fig. 2. It should be noted that the existence of the cross-linking agent DVB would also facilitate the formation of –SO3H groups on the surfaces of LCPS microspheres during the sulfonation reaction, which has been testified by a lot of research.31–33
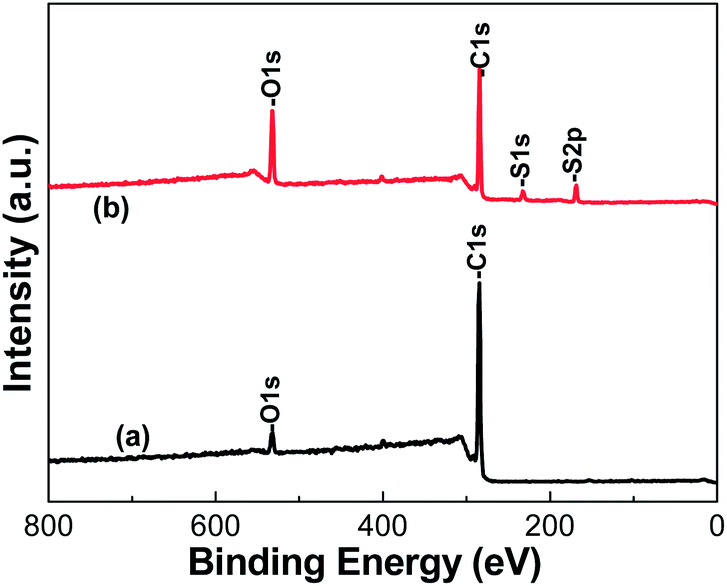 | ||
| Fig. 3 XPS scans of (a) LCPS microspheres and (b) SPS microspheres. | ||
The SPS microspheres with –SO3H groups were subsequently modified with Sn2+ ions to obtain Sn2+-sensitized SPS microspheres. When the acidic SnCl2 aqueous solution was added into the SPS dispersion, the Sn2+ ions were easily adsorbed onto the surfaces of the SPS microspheres via the electrostatic attraction between the negative charged –SO3H groups and positive charged Sn2+ ions (Scheme 1c). The sensitized SPS dispersion was centrifuged to remove unabsorbed Sn2+ ions and redispersed into water. Then, fresh [Ag(NH3)2]+ complex ions aqueous solution and C4H4O6KNa aqueous solution was sequentially poured into the dispersion mentioned above. [Ag(NH3)2]+ ions were in situ reduced to Ag nanoparticles and deposited on the surfaces of modified PS microspheres forming PS/Ag core–shell microspheres (Scheme 1d). Compared with the traditional electroless plating method, this improved process is simpler and milder. In this case, the Sn2+ ions played an important role for the following homogeneous formation process of Ag nanoparticles on the surfaces of PS templates. More specifically, after the Sn2+-sensitized SPS microspheres were mixed with [Ag(NH3)2]+ complex ions, some of [Ag(NH3)2]+ ions were quickly reduced to Ag nanoparticles by Sn2+ ions and fixed on the surfaces of PS microspheres. These initially formed Ag nanoparticles would be served as seeds and promote the later growth of Ag nanoparticles. Then, the residual [Ag(NH3)2]+ ions were further in situ reduced by reducing agent C4H4O6KNa, gradually forming homogeneous and compact Ag nanoparticles on the surfaces of PS microspheres.
The typical SEM images of the large micron-sized PS/Ag core–shell hybrid microspheres shown in Fig. 4a and b indicate that the surfaces of the resulted microspheres become rather rougher as compared with the original LCPS microspheres (Fig. 1) and SPS microspheres (Fig. S1†), and the Ag nanoparticles with an average diameter ∼50 nm are continuously, completely and homogeneously distributed on the surfaces of PS microspheres forming a compact Ag layer, which also hints that the former –SO3H groups and Sn2+ ions are uniformly attached on the surfaces of the micron-sized PS spheres. The TEM images at different magnifications of the typical PS/Ag microspheres are depicted in Fig. 4c and d. As shown in the low magnified TEM image inserted in Fig. 4c, due to the highly compact Ag nanoparticles deposited on the PS microspheres, the PS/Ag hybrid microspheres looks very dark and doesn't show the obvious core–shell structure. In order to survey the microstructure of the surface of the PS/Ag hybrid microsphere, the edge of the microsphere was further magnified and showed in Fig. 4c. It shows that Ag nanoparticles (just as arrow pointed out in Fig. 4c) with a size about 55 nm were grown on the PS surface in granular shape, which is consistent with the SEM image (Fig. 4b). The Ag nanoparticles were tightly bonded to the surface of PS microspheres, indicating the strong adhesion between PS and Ag nanoparticles. However, due to the existence of amorphous structure PS, the edge of Ag nanoparticles and the interface Ag nanoparticles are a little obscure. In addition, in Fig. 4d, the clear lattice fringes marked by the white lines and the measured lattice d-spacing are 0.24 nm, which are very consistent with the interplane distance values of (111) in the face structure Ag crystal, and also indicates the high-crystalline of the in situ generated Ag nanoparticles. The as-prepared PS/Ag bead is a classical core–shell structure of polymer–metal composites and still kept good spherical shape. Due to the existence of Ag nanoparticles layer coating on the surfaces of modified PS microspheres, the diameter of PS/Ag core–shell microspheres increase to about 6 μm. It also can be noticed that each PS microsphere contains immobilized Ag nanoparticles, and no dissociative Ag nanoparticles are found. The micron-sized PS/Ag composites were also synthesized in the absence of Sn2+ ions as a control experiment and the SEM images are shown in Fig. S2.† It can be seen that lots of Ag particles even Ag flake with large and nonuniform size, ranging from 40 nm to 250 nm are loosely piled up on the surfaces of PS microspheres, and a large amount of isolated Ag particles and visible aggregations were formed. The results clearly indicate that the presence of Sn2+ ions would protect the Ag nanoparticles from aggregation during the reduction of [Ag(NH3)2]+ complex ions and facilitate the formation of uniform and compact coverage of Ag nanoparticles on the surfaces of micron-sized PS spheres.
 | ||
| Fig. 4 SEM images (a and b) and TEM images (c and d) of the typical micron-sized PS/Ag core–shell microspheres prepared in the presence of Sn2+ ions. | ||
The XRD patterns of the LCPS microspheres and the corresponding PS/Ag microspheres are illustrated in Fig. 5, which shows that the LCPS microspheres are amorphous corresponding to the broad reflection peak at 2θ = 18.5°. On the contrary, XRD of the PS/Ag hybrid microspheres shows crystalline peaks, where sharp peaks appear at 2θ values of about 38.04°, 44.24°, 64.38°, 77.32° and 81.46°, corresponding to (111), (200), (220), (311) and (222) Bragg's reflections of the fcc structure of Ag (JCPDS no. 04-0783). The disappearance of a strong reflection at 2θ = 18.5° of PS/Ag microspheres illustrates the high and complete coverage of the Ag nanoparticles coating on the surfaces of PS spheres. It confirmed that Ag nanoparticles with good crystallinity had been successfully deposited onto the surfaces of PS cores by reducing [Ag(NH3)2]+ complex ions using Sn2+ ions and C4H4O6KNa.
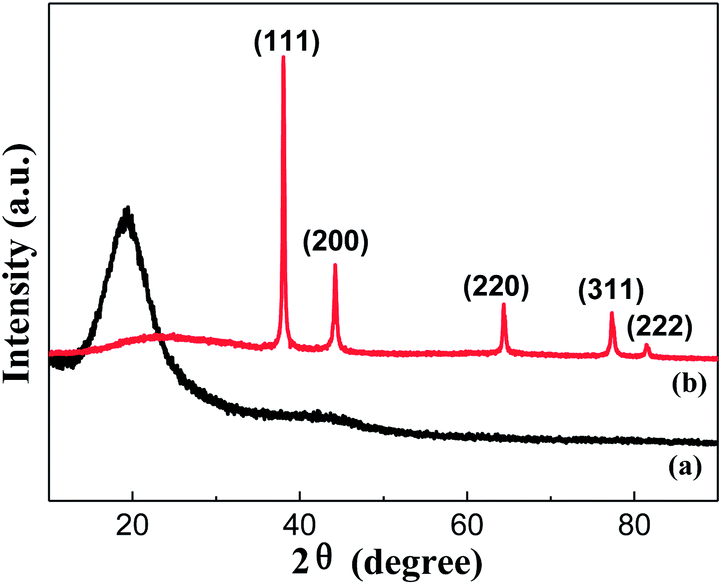 | ||
| Fig. 5 XRD patterns of (a) LCPS microspheres and (b) PS/Ag core–shell microspheres. | ||
3.2. Effect of the concentrations of [Ag(NH3)2]+ complex ions
The concentration of silver precursor, namely [Ag(NH3)2]+ complex ions plays a vital role in the morphology and size control of the target hybrid microspheres. As just mentioned above, Sn2+-sensitized SPS microspheres enable the in situ reduction of [Ag(NH3)2]+ ions to Ag nanoparticles in the existence of C4H4O6KNa, and fixed them on the surfaces of PS microspheres. Fig. 6a–f show SEM images of the PS/Ag microspheres prepared with different concentration of [Ag(NH3)2]+ ions (the concentration of C4H4O6KNa was kept constant as 20 mM) at different magnifications. When the concentration of [Ag(NH3)2]+ ions used in reaction solution was low (15 mM), Ag nanoparticles with average diameter ∼40 nm were coated on the PS beads, and an incomplete nanoshell was formed because of the insufficient amount of silver precursor (Fig. 6a and b). However, when increasing the concentration of [Ag(NH3)2]+ ions in the reaction solution to 30 mM, uniform Ag nanoparticles with bigger size of ∼60 nm and well-shape were deposited completely, continuously and compactly on the surfaces of each PS microsphere, and few free Ag nanoparticles were seen, as shown in Fig. 6c and d. As further increasing the concentration of [Ag(NH3)2]+ ions as high as to 60 mM, the amount of Ag grains immobilized on the PS surfaces increased remarkably, as shown in Fig. 6e and f. Although the Ag coating shell is also complete, the size of the Ag particles is nonuniform, ranging from about 50 nm to 150 nm. Furthermore, it can be seen that some sporadic Ag granules are heaped on the surfaces of the composite microspheres. These phenomena can be explained as follows, the [Ag(NH3)2]+ complex ions were primarily reduced by Sn2+ ions to zero-valent silver and deposited on the surfaces of Sn2+-sensitized SPS microspheres and used as the nucleation center for the formed Ag nanoparticles afterward. Reduced silver species in solution are subsequently deposited to silver nucleation center onto the surfaces of PS microspheres with the aid of a weak reducing agent of C4H4O6KNa. Because the –SO3H groups and Sn2+ ions are uniformly and homogeneously attached on the surfaces of the micron-sized PS spheres, silver nanoparticles nuclei can be homogeneously distribute on the surfaces of PS microspheres, which lead to the following formation of homogeneous and uniform Ag nanoparticles on the PS microspheres. The more [Ag(NH3)2]+ ions attracted onto the micron-sized modified PS spheres, the more the number of resulted Ag nanoparticles formed. Moreover, since the amount of nucleation center kept unchanged, with the concentration of [Ag(NH3)2]+ ions increasing, the size of Ag nanoparticles increased accordingly. | ||
| Fig. 6 SEM images of PS/Ag core–shell microspheres obtained with various [Ag(NH3)2]+ concentrations: (a and b) 15 mM; (c and d) 30 mM; (e and f) 60 mM. | ||
When the concentration of [Ag(NH3)2]+ ions is too high, the reduction rate of Ag(NH3)2+ ions speed up signally, leading to an disorder growth of Ag nanoparticles with nonuniform size. The results mentioned above suggest that Ag nanoparticles with uniform size can be deposited completely and closely on the surfaces of the large micron-sized modified PS spheres resulting in PS/Ag core–shell microspheres by adjusting the concentration of [Ag(NH3)2]+ ions.
In order to obtain large micron-sized hollow metallic spheres, the typical hybrid microspheres of PS/Ag-30 were dissolved by DMF to remove the PS cores. Fig. 7 presents the different magnitude SEM and TEM images of the hollow Ag spheres. As shown in the low magnification SEM image (Fig. 7a), large micron-sized hollow silver shells were obtained and most of the silver shells still retain their stable spherical shape after dissolution the inner polymer core, further indicating that the Ag nanoparticles are homogeneously deposited on the surfaces of PS microspheres to form a continuous, close-packed and stable core–shell structures. The broken silver hollow microspheres might be ascribed to the ultrasonic treatment in the washing process of the as-prepared hollow Ag spheres. The relatively stable hollow structure of the Ag spheres can also be confirmed by the low magnification TEM images (Fig. 7c). Compared with the PS/Ag microspheres (Fig. 4c), the TEM image of the hollow Ag spheres is less dark, indicating that the PS cores were successful removed. Moreover, the magnified TEM image (Fig. 7d) clearly reveals that the hollow Ag spheres are composed of amounts of Ag nanoparticles, and the Ag nanoparticles are contact with their neighbouring particles (circled in Fig. 7d), and it is why these large micron-sized hollow Ag spheres could maintain their relatively stable spherical structure. The inference of the PS cores were completely dissolved by DMF has been confirmed by the result of nearly no weight loss of the hollow Ag spheres from room temperature to 800 °C in the TGA curve, as shown in Fig. 8e.
 | ||
| Fig. 7 SEM (a and b) and TEM (c and d) images of the hollow Ag spheres with different magnifications. | ||
 | ||
| Fig. 8 TGA curves of (a) LCPS microspheres; (b), (c) and (d) PS/Ag-15, PS/Ag-30 and PS/Ag-60, respectively; (e) hollow Ag spheres prepared by dissolving PS/Ag-30 core–shell microspheres. | ||
The thickness of silver shells is estimated to be ∼110 nm as shown in the high magnification SEM image (Fig. 7b), which is remarkably close the theoretical simulation value (please refer to “Estimation of the Ag shell thickness in the PS/Ag core–shell microspheres from TGA data” of ESI†). It is amazing that the Ag shells could keep their spherical morphology with so large size (∼6 μm), which is rare reported in the previous reports. The hollow Ag spheres can be used as low density conducting materials, catalysts, drug release agents, and gas storage materials and are the subject of a future work. Therefore, all the evidence demonstrates that the strategy is succeed in preparation of the large micron-sized monodisperse PS/Ag spheres with compact and complete Ag nanoparticles shells using an improved electroless plating method, and the corresponding micron-sized hollow Ag spheres are easily obtained via a simple dissolution process of the as-synthesized PS/Ag core–shell microspheres. Compared with traditional electroless plating method, the whole procedure is mild, efficient and eco-friendly.
TGA was used to study the thermal stability of various as-prepared particles (LCPS microspheres, PS/Ag composite microspheres and hollow Ag spheres) and calculate the corresponding weight percentage of Ag nanoparticles in the composites, as shown in Fig. 8. TGA curve of the LCPS (Fig. 8a) illustrates that the pure LCPS microspheres start to lose weight at around 300 °C, and almost completely decomposes at temperature above 470 °C. Although, in principle, the LCPS should be converted to carbon under anaerobic circumstances at a high temperature (800 °C), a tiny amount of oxygen would help to burn them away. Thus, the residual weight should be that of silver in the cures of Fig. 8b–d. According to the TGA curves, the residue silver contents of samples PS/Ag-15, PS/Ag-30 and PS/Ag-60 are found to be 45.17%, 55.03% and 60.77%, respectively. The results show that, as [Ag(NH3)2]+ ions concentration increasing, the more silver particles are loaded on the PS microspheres, the more residues are left over, which is consistent with the SEM results shown in Fig. 6. The thermal stability of samples was also observed from the TGA curves. The onset temperature of degradation (the temperature of 5% weight loss) of the pristine LCPS microspheres was 334 °C. After the LCPS microspheres were coated by silver particles, all PS/Ag samples exhibit a major increase in the onset temperature of degradation relative to virgin LCPS, which indicates an enhancement in the thermal stability as a result of the protection layer of Ag nanoparticles. In detail, the onset temperature of degradation of PS/Ag-15, PS/Ag-30 and PS/Ag-60 hybrid microspheres was 393 °C, 398 °C and 401 °C, respectively. All the above results demonstrate that with increasing the concentration of [Ag(NH3)2]+ ions, the content of Ag nanoparticles loaded on PS spheres and the heat resistance of the as-prepared PS/Ag hybrid microspheres are increased.
3.3. The electrical properties of monodisperse PS/Ag core–shell microspheres
The bulk electrical conductivity of the as-synthesized large micron-sized PS/Ag core–shell hybrid spheres was investigated via a four-point probe system so as to evaluate the potential use of this material in anisotropic conductive adhesives. The bulk electrical conductivity of the as-prepared PS/Ag-15 containing 45.17% silver is 3.76 × 104 S m−1. As the concentration of [Ag(NH3)2]+ ions increase to 30 mM, the bulk electrical conductivity of the PS/Ag-30 containing 55.03% silver is drastically increased to 2.0 × 105 S m−1. When the concentration of [Ag(NH3)2]+ ions increase as high as 60 mM, the bulk electrical conductivity of the resulting PS/Ag-60 containing 60.77% silver reach up to 3.33 × 105 S m−1. The results show that with the increase of the concentration of [Ag(NH3)2]+ ions in reaction solution, the electrical conductivity of the resulting PS/Ag core–shell microspheres increases. This can be explained as follows. The micron-sized PS/Ag core–shell spheres with a relatively low silver content as 45.17%, silver coating was incomplete as shown in Fig. 6a and b, resulting in a poor contact between PS/Ag microspheres. When the silver content of PS/Ag microspheres is 55.03% or higher, continuous and close-packed silver shells were completely formed, as shown in Fig. 6c–f. So the Ag nanoparticles are touching each other easily, leading to a higher bulk electrical conductivity value, which suggest that the as-prepared PS/Ag core–shell composite microspheres could be used as ideal conductive fillers. It is worth mentioning that the electrical conductivity of the as-prepared monodisperse PS/Ag microspheres with a diameter ca. 6 μm is much higher than the optimal value of 103 S m−1 and 3.57 × 104 S m−1 reported by Ma et al.13 and Wang et al.,20 respectively. These excellent results can be attributed to the high silver content of PS/Ag composite microspheres, uniform and compact coverage and no any organic matters (e.g. PVP, glucose) were used in the reduction of Ag nanoparticles onto the surface of PS cores. As is known to all, the remaining of organic matters on the surfaces of metal particles will decrease their electrical properties.3.4. The catalytic activities of monodisperse PS/Ag core–shell microspheres and hollow Ag spheres
Metallic silver nanoparticles have been extensively investigated for their catalytic activities for degradation of nitrophenols, nitroanilines and various dyes, such as methylene blue (MB) and fluorescein.34,35 Dyes are a major class of synthetic organic compounds released by many industries, and result in significant environmental pollution. Unfortunately, their catalytic abilities would be decreased or even lost due to the easily aggregation of metallic nanoparticles. Coating silver nanoparticles onto the surfaces of some large-sized particle templates is an effective way to solve this problem. Herein, the catalytic activities of the large micron-sized monodisperse PS/Ag core–shell microspheres and the hollow Ag spheres were examined using the reduction of dye MB by NaBH4 as a representative reaction. MB can be reduced by reductants like NaBH4 to form leucomethylene blue (LMB), but the reduction rate is very slow.36 Metal nanoparticles with high reactive activity and specific surface area could accelerate the reduction rate of dyes, thus increasing the reducing efficiency. The as-synthesized micron-sized PS/Ag-30, PS/Ag-60 and hollow Ag spheres, corresponding to Fig. 6c, e and 7, respectively, are selected as catalysts. The reduction process was monitored with a UV-Visible spectroscopy. The absorbance maximum (λmax) of MB monomer in water appears normally at 665 nm, corresponding to the n–π* transition and a shoulder peak at 614 nm.37–39 The time-dependent UV-Vis absorption spectra are shown in Fig. 9. The decreasing trend of the absorption intensity indicates the gradually reduction of MB by NaBH4. From Fig. 9a, it is known that, without adding the catalysts, the UV-Vis absorbance at λmax of MB decrease from 1.503 to 0.951 within 90 min in a slow pace. Increased degradation velocity of MB has been achieved through the addition of Ag catalysts which is shown by strong decrease in the absorption intensity (Fig. 9b–e). When 0.2 mL PS/Ag-30 dispersion was added into the reaction solution, the complete reduction of MB is accomplished in a period of 25 min (Fig. 9b). For the same volume of PS/Ag-60 catalysts was introduced, the complete degradation of MB is finished less than 11 min (Fig. 9c). Namely, a higher content of Ag nanoparticles loaded on the surfaces of PS microspheres and larger specific surface area (listed in Table 1 and the original data shown in Fig. S4†) is found to be further facilitated the reduction process of MB to LMB. Surprisingly, the absorbance of MB at λmax decrease dramatically to zero in only 3 min in the presence of 0.2 mL hollow Ag spheres dispersion (Fig. 9d). In view of the fact that the actual Ag content of the hollow Ag spheres dispersion is higher than that of the same volume PS/Ag-30 dispersion, a lower volume of 0.11 mL hollow Ag spheres dispersion (with this volume, the actual Ag mass content in the catalytic system is same as the PS/Ag-30 catalysts) was used in the reaction solution to further evaluate their catalytic activities. As shown in Fig. 9e, the UV-Vis absorption peak at λmax also decreases rapidly and the complete disappearance of MB appears at 7 min. The time of reaction completely is markedly shorter than that of using 0.2 mL PS/Ag-30 dispersion as catalysts. | ||
| Fig. 9 Time-dependent UV-Vis absorption spectra for the reduction of MB by NaBH4 (a) without catalyst; (b–e) with 0.2 mL PS/Ag-30, 0.2 mL PS/Ag-60, 0.2 mL hollow Ag spheres and 0.11 mL hollow Ag spheres, respectively; (f) the natural logarithm of the absorbance at λmax versus the reduction time. | ||
| No. | Catalyst | Volume (mL) | Ag content in the catalytic system (mg mL−1) | BET (m2 g−1) | k for MBa (min−1) |
|---|---|---|---|---|---|
| a k: first-order reaction rate constant. | |||||
| 1 | No catalyst | — | — | — | 0.00488 |
| 2 | PS/Ag-30 | 0.2 | 0.011 | 2.59 | 0.13567 |
| 3 | PS/Ag-60 | 0.2 | 0.012 | 3.03 | 0.31661 |
| 4 | Hollow Ag spheres | 0.2 | 0.02 | 4.68 | 1.97857 |
| 5 | Hollow Ag spheres | 0.11 | 0.011 | 4.68 | 0.73141 |
The relative absorbance of band at λmax is plotted as a function of time to establish reaction kinetics and evaluate the reduction reaction rate. In our experiments, the initial concentration of NaBH4 (25 mM) greatly exceed that of MB (0.05 mM), so we consider the decrease of the concentration of NaBH4 is negligible during the reaction. In other words, the reduction of MB by NaBH4 follows pseudo-first order reaction kinetics.34,35,40 The pseudo-first order rate is based on the equation ln(At/A0) = kt, where A0 and At are the absorbance value of MB initially and at the reaction time t (min), respectively, and k is the apparent first-order rate constant (min−1). The plots of ln(At/A0) versus time t are shown in Fig. 9f. The plots are good linear correlation with the reaction time and the apparent first-order rate constant k could be directly obtained from the slope of the linear plots and given in Table 1. For the contrast experiment without catalysts, the value of k is very low (0.00488 min−1), intuitively reflecting that the degradation of MB only by NaBH4 is difficult and time-consuming. Then, the rate constant k increases to 0.13567 min−1 with 0.2 mL PS/Ag-30 dispersion as the catalyst, which indicates the obvious catalytic effect. Moreover, for the catalytic system supported with 0.2 mL PS/Ag-60 dispersion, the value of k is 0.31661 min−1, which is more than twice that of the system assisted with 0.2 mL PS/Ag-30. The increase of the rate constant directly indicates the speeding up of the degradation of the dye. Furthermore, the rate constant k signally increases to 0.73141 min−1 (for 0.11 mL hollow Ag spheres) and 1.97857 min−1 (for 0.2 mL hollow Ag spheres), respectively, which is several times larger than that of PS/Ag-30.
The main reason for the excellent catalytic activity is associated with the special structure of catalysts and can be explained by the mechanism of the catalytic reaction. NaBH4 is ionized in water to offer BH4−, which provide surface hydrogen for the reaction. The surface hydrogen is first transferred to Ag nanoparticles, and then reacted with MB to yield LMB. BH4− acts as the electron donor, while MB acts as the electron acceptor. Ag nanoparticles could help the electron to transfer from the donor to the acceptor. Due to the catalysis occurs only on the surface of metal particles, so particles that possess high surface areas might act as good substrates for the electron transfer reaction. Therefore, increasing the available surface area will greatly enhance the effectiveness of the catalyst. As mentioned above, the as-prepared PS/Ag microspheres own typical core–shell structure and compact Ag nanoparticles are distributed uniformly on the PS microsphere surface. This core–shell structure could effectively avoid the aggregation of Ag nanoparticles and supply a high surface area substrate. When the inner polymer cores are removed and forming the corresponding hollow Ag spheres, the spherical shape of catalysts are generally remained, and the contact area between the catalysts and reaction solution is increased. It also proven that the BET specific surface area of the hollow Ag spheres is higher than that of PS/Ag-30 (Table 1). Thus, it is believed that the as-prepared core–shell PS/Ag composite microspheres and their corresponding hollow Ag spheres is a promising catalyst for the reduction of dyes.
4. Conclusions
In summary, we present a simple, efficient and environmentally friendly method to prepare large micron-sized monodisperse PS/Ag core–shell hybrid microspheres with a continuous, complete and compact silver nanoparticles layer by an improved electroless plating process. Then, the corresponding micron-sized hollow Ag spheres could be easily obtained via simply dissolving of the inner PS cores by solvent. FTIR, XPS, XRD, TGA and SEM results have demonstrated the formation process, morphology and structure of the as-prepared microspheres. Moreover, the size and morphology of the Ag nanoparticles on the surfaces of the monodisperse LCPS microspheres can be facilely tuned by altering the concentration of the silver precursor. The electrical conductivity of the as-prepared PS/Ag composites increased from 3.76 × 104 S m−1 to 3.33 × 105 S m−1, corresponding to a silver content of 45.17–60.77%, which suggest that monodisperse PS/Ag core–shell microspheres could be used as ideal conductive fillers for the anisotropic conductive adhesives. Moreover, the as-prepared micron-sized PS/Ag core–shell spheres exhibit excellent activity in the catalytic reduction of MB by NaBH4. In particular, the hollow Ag spheres demonstrate more outstanding catalytic activity due to their stable hollow structure and higher specific surface area. Experimental results show that the rate constant k with the assistance of hollow Ag spheres is several times that obtained by the support of PS/Ag microspheres.Furthermore, this eco-friendly method based on modified electroless plating also present a new paradigm to deposit different metallics (Au, Cu, Pt, and Ni, etc.) onto various polymer material surfaces. On basis of the technique, many kinds of materials coated with various metal nanoparticles could be achieved, which are expected to be promising nanocomposite materials for optical, magnetic, anti-bacterial, electrical conductive and catalytic applications.
Acknowledgements
The authors are grateful for the financial support from the National Basic Research Program of China (973 Program) (2012CB933700-G), National Natural Science Foundation of China (21101165), Guangdong Innovative Research Team Program (no. 2011D052), Shenzhen Peacock Planning Team (KYPT20121228160843692), Shenzhen Electronic Packaging Materials Engineering Laboratory (2012-372). Shenzhen basic research plan (JC201005270372A and GJHS20120702091802836).Notes and references
- D. Lu and C. P. Wong, Advanced Flip Chip Packaging, ed. H. M. Tong, Y. S. Lai and C. P. Wong, 2013, pp. 201–261 Search PubMed.
- H. V. Nguyen, A. Erik, K. Helge, J. Rolf, N. Hoivik and K. E. Aasmundtveit, Mater. Des., 2013, 46, 784–793 CrossRef CAS PubMed.
- Y. G. Hu, T. Zhao, P. L. Zhu and R. Sun, Colloid Polym. Sci., 2012, 290, 401–409 CAS.
- Y. X. Li, Y. Wu, Y. Gao, S. S. Sha, J. F. Hao, G. Q. Cao and C. Yang, RSC Adv., 2013, 3, 26361–26366 RSC.
- F. Wen, W. Q. Zhang, G. W. Wei, Y. Wang, J. Z. Zhang, M. C. Zhang and L. Q. Shi, Chem. Mater., 2008, 20, 2144–2150 CrossRef CAS.
- K. Kim, H. B. Lee, H. K. Park and K. S. Shin, J. Colloid Interface Sci., 2008, 318, 195–201 CrossRef CAS PubMed.
- O. Siiman and S. Ledis, J. Raman Spectrosc., 2005, 36, 1125–1133 CrossRef CAS.
- P. Tierno and W. A. Goedel, J. Phys. Chem. B, 2006, 110, 3043–3050 CrossRef CAS PubMed.
- G. Saini, D. S. Jensen, L. A. Wiest, M. A. Vail, A. Dadson, M. L. Lee, V. Shutthanandan and M. R. Linford, Anal. Chem., 2010, 82, 4448–4456 CrossRef CAS PubMed.
- Y. Cong, T. Xia, M. Zou, Z. N. Li, B. Peng, D. Z. Guo and Z. W. Deng, J. Mater. Chem. B, 2014, 2, 3450–3461 RSC.
- N. Kawahashi and H. Shiho, J. Mater. Chem., 2010, 10, 2294–2297 RSC.
- Z. H. Yang, L. L. Yang, Z. F. Zhang, N. Z. Wu, J. L. Xie and W. X. Cao, Colloids Surf., A, 2008, 312, 113–117 CrossRef CAS PubMed.
- Y. H. Ma and Q. H. Zhang, Appl. Surf. Sci., 2012, 258, 7774–7780 CrossRef CAS PubMed.
- X. J. Cheng, S. C. Tjong, Q. Zhao and R. K. Y. Li, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4547–4554 CrossRef CAS.
- H. X. Guo, Z. P. Qin, J. Wei and C. X. Qin, Surf. Coat. Technol., 2005, 200, 2531–2536 CrossRef CAS PubMed.
- S. C. Tang, L. Chen, S. Vongehr and X. K. Meng, Appl. Surf. Sci., 2010, 256, 2654–2660 CrossRef CAS PubMed.
- W. J. Cai, W. Q. Wang, Y. Q. Yang, G. H. Ren and T. Chen, RSC Adv., 2014, 4, 2295–2299 RSC.
- A. G. Dong, Y. J. Wang, Y. Tang, N. Ren, W. L. Yang and Z. Gao, Chem. Commun., 2002, 350–351 RSC.
- J. H. Lee, J. S. Oh, P. C. Lee, D. O. Kim, Y. Lee and J. D. Nam, J. Electron. Mater., 2008, 37, 1648–1652 CrossRef CAS PubMed.
- W. C. Wang, Y. Jiang, S. P. Wen, L. Liu and L. Q. Zhang, J. Colloid Interface Sci., 2011, 368, 241–249 CrossRef PubMed.
- P. Ritwik and S. S. Kumar, RSC Adv., 2013, 3, 7808–7815 RSC.
- P. Tierno and W. A. Goedel, J. Phys. Chem. B, 2006, 110, 3043–3050 CrossRef CAS PubMed.
- H. Mackova, F. Oukacine, Z. Plichta, M. Hruby, J. Kucka, M. Taverna and D. Horak, J. Colloid Interface Sci., 2014, 421, 146–153 CrossRef CAS PubMed.
- M. Okubo, E. Ise and T. Yamashita, J. Appl. Polym. Sci., 1999, 74, 278–285 CrossRef CAS.
- H. F. Qi, W. C. Hao, H. Z. Xu, J. Y. Zhang and T. M. Wang, Colloid Polym. Sci., 2009, 287, 243–248 CAS.
- K. C. Lee and H. A. Wi, J. Appl. Polym. Sci., 2010, 115, 297–307 CrossRef CAS.
- J. S. Song and M. A. Winnik, Macromolecules, 2005, 38, 8300–8307 CrossRef CAS.
- M. L. Cao, B. Tong, J. B. Shen, Y. P. Dong and J. G. Zhi, J. Appl. Polym. Sci., 2008, 109, 1189–1196 CrossRef CAS.
- N. Mariagrazia, D. Christophe, I. Ivano, L. Pasquale and G. Gaetano, Macromolecules, 2009, 42, 5566–5571 CrossRef.
- Y. Yang, Y. Chu, F. Y. Yang and Y. P. Zhang, Mater. Chem. Phys., 2005, 92, 164–171 CrossRef CAS PubMed.
- A. M. Muhammad, Ind. Eng. Chem. Res., 2009, 48, 6961–6965 CrossRef.
- A. Linares and J. L. Acosta, Macromol. Mater. Eng., 2005, 290, 53–59 CrossRef CAS.
- O. Ozlem, I. Ahmet, K. Bunyamin and B. Niyazi, Desalination, 2013, 309, 141–147 CrossRef PubMed.
- M. Wang, D. Tian, P. P. Tian and L. J. Yuan, Appl. Surf. Sci., 2013, 283, 389–395 CrossRef CAS PubMed.
- E. Murugan and J. N. Jebaranjitham, Chem. Eng. J., 2015, 259, 266–276 CrossRef CAS PubMed.
- V. K. Vidhu and D. Philip, Micron, 2014, 56, 54–62 CrossRef CAS PubMed.
- Y. Zhang, P. L. Zhu, L. Chen, G. Li, F. R. Zhou, D. Lu, R. Sun, F. Zhou and C. P. Wong, J. Mater. Chem. A, 2014, 2, 11966–11973 CAS.
- M. K. Mohammad, J. Lee and M. H. Cho, J. Ind. Eng. Chem., 2014, 20, 1584–1590 CrossRef PubMed.
- V. S. Suvith and D. Philip, Spectrochim. Acta, Part A, 2014, 118, 526–532 CrossRef CAS PubMed.
- M. Y. Tang, X. R. Wang, F. Wu, Y. Liu, S. Zhang, X. B. Pang, X. X. Li and H. X. Qiu, Carbon, 2014, 71, 238–248 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: A SEM image of SPS microspheres; SEM images of PS/Ag hybrid microspheres prepared without Sn2+ ions; estimation of the Ag shell thickness in the PS/Ag core–shell microspheres from TGA data; BET figure from nitrogen adsorption isotherms. See DOI: 10.1039/c4ra12475h |
| This journal is © The Royal Society of Chemistry 2015 |