
Advanced construction of heterostructured InCrO4–TiO2 and its dual properties of greater UV-photocatalytic and antibacterial activity
Abstract
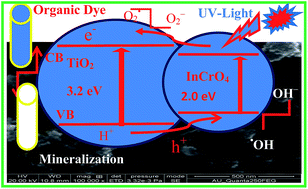
The development of coupled semiconductor oxides has led to significant advances in photocatalytic functional materials. In this article, we report the preparation of a nano-spherical InCrO4-loaded TiO2 photocatalyst by a simple co-precipitation method. The InCrO4–TiO2 (ICT) nanomaterial was characterized using XRD, SEM with EDX, FT-IR, FT-Raman and PL analysis. The photocatalytic activity of InCrO4–TiO2 was much better than that of TiO2 and TiO2–P25 under UV-light irradiation. InCrO4–TiO2 showed better activity than TiO2 and TiO2–P25 in the degradation of methyl green (MEG), malachite green (MAG) and methylene blue (MB) because it has maximum efficiency at neutral pH = 7 under UV-light irradiation. Among all these dyes, the photodegradation of MEG was the fastest. The mechanism for the photocatalytic activity of the InCrO4-loaded TiO2 nanomaterial has been discussed. The production of hydroxyl radicals on the surface of UV-irradiated photocatalysts was determined by fluorescence technique using coumarin as a probe molecule. The photodegradation of dyes is well described by pseudo first order kinetics and high quantum yield. Furthermore, the antibacterial activity of the TiO2 and InCrO4–TiO2 materials was investigated against Gram negative Vibrio cholerae and Gram positive Bacillus subtilis bacterial strains.
 Please wait while we load your content...
Please wait while we load your content...

