
Development of a highly efficient single-mode microwave applicator with a resonant cavity and its application to continuous flow syntheses†
Abstract
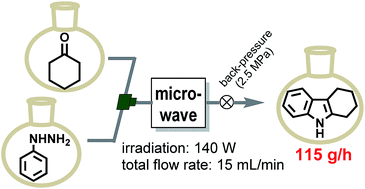
Microwave-assisted organic synthesis has many advantages and is widely applied to a variety of reactions. We have developed a new single-mode microwave applicator, specific to continuous flow synthesis, whose main feature is that it generates a uniform electromagnetic field inside its resonant cavity. Two well-known reactions, the Fischer indole synthesis and the Diels–Alder reaction, proceeded very quickly when a solution of substrates was pumped through a helical glass tube reactor inside the resonant cavity, under a pressure of 2.5 MPa. The desired products were obtained in high yields. This compact apparatus constitutes a new method for switching organic synthesis from batch to continuous flow, and enables continuous synthesis of products at a scale of 100 g h−1 or more.
 Please wait while we load your content...
Please wait while we load your content...

