DOI:
10.1039/C4RA12356E
(Paper)
RSC Adv., 2015,
5, 6443-6451
Electrochemical performance of reduced graphene oxide surface-modified with 9-anthracene carboxylic acid†
Received
14th October 2014
, Accepted 17th December 2014
First published on 17th December 2014
Abstract
An efficient approach for the preparation of 9-anthracene carboxylic acid (ACA) modified reduced graphene oxide (rGO) was demonstrated in this study. ACA was used as a surface-modifying agent and underwent a reversible redox reaction. The benzene ring of the ACA anion was attached to the rGO surface via π–π interactions, and the carboxylate anions helped to disperse the hybrid materials in water due to hydrogen bonding. Therefore, water-dispersible, ACA-modified rGO (ACA-rGO) improved the wettability and capacitance performance in aqueous electrolyte solutions. The morphology of the ACA-rGO was studied using transmission electron microscopy and atomic force microscopy image analysis. The dispersion characteristics of the exfoliated materials were investigated using UV-vis spectroscopy analysis. The chemical states and natures of the samples were investigated using Fourier-transform infrared spectroscopy and X-ray photoelectron spectroscopy (XPS). The appearance of a new peak at 288.7 eV in the XPS of ACA-rGO confirmed the successful surface modification of rGO using ACA. Raman spectra were studied to compare the electronic structure and defect concentrations in the ACA-rGO with respect to GO. The low intensity and shifted D- and G-bands indicated non-covalent functionalization of rGO with ACA anions. Electrochemical performances of ACA-rGO, rGO, and GO were evaluated in 1 M aqueous Na2SO4 electrolyte. The capacitance performance was investigated through galvanometric charge–discharge with ACA-rGO, rGO, and GO in an operating voltage of −1 to 1 V. The range of specific capacitance in the three-electrode system was 610 to 115 F g−1 at a current density range of 0.8 to 20 A g−1. In addition, the capacitance performance of ACA-rGO was studied in 1 M Na2SO4 electrolyte using two-electrode systems. The cell capacitance, energy density, and power density at a current density of 0.2 A g−1 of the asymmetric assembly with multiwall carbon nanotubes were 77 F g−1, 41.3 Wh kg−1, and 200 W kg−1, respectively.
1. Introduction
Continual research on energy-intensive materials is necessary to improve the performance of supercapacitor or electrochemical capacitor (EC) devices due to the increasing demand for energy storage devices for microelectronics and hybrid vehicles. Supercapacitors are progressing as energy storage devices as well as high-power supply devices because they exhibit several desirable characteristics in an energy storage system, such as high power density, long cycle stability, fast charging and discharging capability and facile device integration technology. Supercapacitors can store energy through the combination of electrostatic attraction of opposite charges and fast faradic charge transfer reaction between electrolyte and electrode. Commercially available supercapacitors are generated using active carbon black, fullerenes, carbon fiber, and carbon nanotubes.1,2 The energy storage mechanisms in supercapacitor are based on double layer formation at the interface of electrolyte and electrode. The double layer formation again depends on the active surface area, porosity, and wettability of the electrode materials. However, due to the microporosity (<2 nm) of carbon black, the maximum electrode surface is not wetted by electrolytes, which limits the performance of the supercapacitor.3 In contrast, supercapacitor efficiency can be increased through a faradic reaction, which is a mechanism similar to that used in batteries. Therefore, metal oxides, like RuO2, IrO2, manganese, and nickel-based oxides, have been used as electrodes for supercapacitor applications.2 However, metal oxides can be restricted by availability, high cost, limited redox potential voltage range, and poor electrical conductivity. Therefore, the search for electrochemically stable, large surface area, uniformly porous, and high electrically conductive materials continues for the development of next-generation energy storage devices.
The discovery of graphene in 2004 marked a new era in the area of materials research due to its outstanding mechanical, thermal, electrical, and chemical properties.4–8 Graphene is the thinnest possible two-dimensional layer of graphite. It can be prepared by mechanical exfoliation of graphite, heat treatment of silicon carbide, and chemical vapour deposition of hydrocarbon.9 Recently, graphene has been of high interest in the area of energy storage. However, the limited wettability with aqueous and non-aqueous solvents and restacking of individual graphene sheets are the major concerns for its use as an electrode material for supercapacitor applications.
In order to overcome these issues, surface modification of graphene has been introduced.9,10 Interestingly, chemical techniques are most effective for the scaled-up production of surface-modified graphene to fulfil the demand for supercapacitor applications. Surface modification of graphene can be achieved through the preparation of composites using metal oxides, small organic molecules, macromolecules, or conducting polymers.10–14 However, metal oxide and conducting polymer-based graphene composites have limited processability. The electron transition after redox reaction is limited by the size and surface morphology of metal oxides and conducting polymers.15,16 In addition, the capacitive behaviour of the metal oxide and conducting polymer-based graphene composites is dominated by a pseudocapacitance component, which is not desirable due to a slow response time and high capacitance decay rate. In contrast, redox active small molecule-modified graphene is helpful for improving the electron transfer between electrode and electrolyte during the charge–discharge process. Moreover, it can improve the wettability via dipolar interactions.11,12
Herein, surface modification of graphene using 9-anthracene carboxylic acid (ACA) has been demonstrated. ACA is a tricyclic hydrocarbon and can serve as a semiconductor due to its extended π-electron cloud.13 The carboxylic functional groups of ACA help to increase the redox reaction and wettability with an aqueous electrolyte. The electrochemical performance of water-dispersible GO, ACA-modified reduced graphene oxide (ACA-rGO) and reduced graphene oxide (rGO) have been investigated in 1 M Na2SO4 aqueous electrolyte to determine their utility as electrode materials for energy storage applications. In addition, multiwall carbon nanotubes (MWNTs) were employed for enhancing the capacitance performance using asymmetric (ACA-rGO/Separator/MWNTs) configuration in 1 M Na2SO4 electrolyte. The MWNTs are attractive material for supercapacitor electrode due to its high electrical conductivity, unique network of mesopores and high electrochemical stability. Wang et al.17 demonstrated that randomly oriented bucky-paper electrode of MWNTs showed higher capacitance value than highly oriented MWNTs bucky-paper electrode. The large surface area and highly porous nature of randomly oriented MWNTs contribute to enhancing the electrochemical performance of a hybrid supercapacitor. Frackowiak et al.18 showed that high specific capacitance of 90 F g−1 was achieved using MWNTs material. Thus, ACA-rGO and MWNTs have been used as negative and positive electrodes for asymmetric system.
2. Experimental studies
2.1 Materials
Natural graphite flakes (Sigma-Aldrich, Steinheim, Germany) were used as received. Sulfuric acid (99%), hydrochloric acid, hydrogen peroxide, sodium hydroxide, and sodium sulfate were purchased from Samchun Pure Chemical Co., Ltd. (Pyeongtaek, Korea). Potassium permanganate (Junsei Chemical Co., Ltd., Japan) was used as an oxidizing agent. The surface modifier (ACA) was purchased from Fluka, Germany.
2.2 Preparation of graphite oxide
Graphene oxide (GO) was prepared according to a method reported earlier.14,17 In brief, about 2 g of natural graphite flakes were dispersed in 46 mL of concentrated sulphuric acid in a 250 mL round bottom flask placed on an ice-bath. Subsequently, ∼6 g of potassium permanganate was added slowly over 15–20 min, and the resulting mixture was stirred for 2 h at a constant temperature of 0–5 °C. The reaction mixture was then transferred to a pre-heated oil bath at 35 °C and was stirred for 6 h. Finally, ∼92 mL of de-ionized (DI) water was carefully added to the reaction mixture, followed by stirring for another 1 h. The whole reaction mixture was then poured into a 1 L beaker containing 240 mL of water. About 35% of a hydrogen peroxide solution was added until the colour changed to bright yellow. Dilute hydrochloric acid (5% in water) was added in order to remove the excess manganese salt. The synthesized GO was suspended in water to give a brown dispersion and was subjected to repeated washing with DI water to completely remove residual salts and acids. The purified GO was then dispersed in water (0.5 mg mL−1) to yield a stable dispersion. GO was exfoliated by ultrasonication using an ultrasonic bath (Sonosmasher ULH700S, 20 kHz).
2.3 Synthesis of ACA-rGO and rGO
About 50 mg of GO was dispersed in 200 mL of DI water by sonication for 30 min in a water bath, after which the solution turned brown. About 100 mg of ACA was dissolved in 50 mL of DI water in the presence of a few drops of a 1 M aqueous NaOH solution. Alkaline 9-ACA was added to the GO supernatant solution. The mixture was stirred for 24 h at 60 °C, and then 0.05 mL hydrazine monohydrate was added to the supernatant mixture, followed by continuous stirring for another 24 h. The final product was washed using vacuum filtration until a pH of 7 was achieved. The washed solution, dubbed ACA-rGO, was used for further characterization. Similarly, rGO was prepared without adding ACA but using the same amount of NaOH.
2.4 Characterization
The aqueous dispersion of ACA-rGO was analysed using UV-visible spectroscopy at ambient temperature using a UVS-2100 SCINCO spectrophotometer. Fourier transform infrared (FT-IR) spectroscopy was performed using a Nicolet 6700 spectrometer (Thermo Scientific, USA) over the wave number range of 4000 to 400 cm−1. Raman spectra of GO and ACA-rGO were obtained on a Nanofinder 30 (Tokyo Instruments Co., Osaka, Japan). The chemical characteristics of graphite, GO, and ACA-rGO were analysed by X-ray photoelectron spectroscopy (XPS) (AXIS-NOVA, Kratos Analytical Ltd., Manchester, UK) using a 160 eV pass energy and a step size of 0.7 eV to ensure the purity and cleanliness of the samples. Pass energy of 50 eV and a step size of 0.1 eV were used for narrow spectra, and the energy resolution was <0.6 eV, which was determined as the full-width-at-half-maximum (FWHM) of the Au4f7/2 line calibrated to 83.9 eV. Atomic force microscopy (AFM) analysis of ACA-rGO was carried out using anXE-100 atomic force microscope from PARK Systems (Suwon, South Korea). AFM samples were prepared by deposition of a water dispersion of functionalized graphene on the surface of a silicon substrate, followed by drying in a N2 atmosphere. Microstructures of the samples were investigated using a JEM-2200 FS transmission electron microscope (TEM) (JEOL, Japan). Uniform aqueous dispersions of functionalized graphene sheets (ACA-rGO) were drop-cast onto a fresh lacey carbon Cu-grid. Thermogravimetric analysis (TGA) was carried out on a Q50TGA (TA Instruments, New Castle, DE, USA) at a heating rate of 5 °C min−1 from 60 to 800 °C in a nitrogen atmosphere.
2.5 Electrochemical measurements
Electrochemical performance was measured using three and two electrodes system on a CH660D electrochemical workstation. The fabrication of working electrodes was accomplished as described elsewhere.19 In brief, the water-dispersible ACA-rGO was slowly casted (3 mg cm−2) onto nickel foam without binder and was dried at 60 °C for 24 h in a vacuum oven. Similarly, for two-electrode studies, raw MWCNTs were fabricated on the nickel foam using a PDVF binder (5%) and were dried under same conditions mentioned above. Then, two electrodes were assembled using a PTEF (25 μm thickness and 2 μm pore size) separator and a 1 M Na2SO4 aqueous solution as an electrolyte. About 3 mg cm−2 mass density was used throughout these studies. Finally, the assembled electrode was set in the sealed electrode chamber. Electrochemical impedance spectroscopy (EIS) was performed at open circuit voltage potential. The specific capacitance (Cs) values were calculated by both ways: (1) Cs = I × Δt/m × ΔV, where I = current, m = mass of deposited material, Δt = discharging time, and ΔV = operating voltage, based on the charge–discharge curve and (2)  , where I = instantaneous current, V1, V2 = cut-off potentials, and μ = potential scan rate, based on the cyclic voltammetry (CV) curve. Energy density (E) and power density (P) were calculated using the respective equations E = (1/2)CV2 and P = E/t.12,20,21
, where I = instantaneous current, V1, V2 = cut-off potentials, and μ = potential scan rate, based on the cyclic voltammetry (CV) curve. Energy density (E) and power density (P) were calculated using the respective equations E = (1/2)CV2 and P = E/t.12,20,21
3. Results and discussions
3.1 TEM and AFM image analysis
TEM and selected area electron diffraction (SAED) pattern images were recorded to determine the morphology of ACA-rGO. Fig. 1(a) and (b) clearly show the existence of thin ACA-rGO sheets on the Cu-grid. In addition, the SAED pattern indicated that the crystalline nature was preserved after functionalization and formation of a single layer ACA-rGO sheet.12,14,22 Atomic force microscopy (AFM) was performed to investigate the topographic height of ACE-rGO. The average thickness of ACA-rGO was 0.897 nm, as shown in Fig. 1(c). Considering the adsorption of ACA on the surface of a graphene sheet, the observed thickness indicates the formation of single-layer ACA-rGO after chemical functionalization.12,14,22
 |
| | Fig. 1 (a) TEM (b) SAED pattern and (c) AFM image of ACA-rGO. | |
3.2 UV-vis spectra analysis
The UV-vis absorption of GO, ACA-rGO, and rGO are depicted in Fig. 2, and the inset picture indicates a visual photograph of dispersion in aqueous medium. The pure GO shows absorption peaks at 230 nm, probably due to the π–π* transition of the C–C bond. Additionally, a shoulder appeared at ∼300 nm due to the n–π* transition in the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond. In contrast, after functionalization and reduction, the peak at 230 nm shifted to ∼256 nm, indicating the reduction of GO. Shoulder-like weak peaks appeared at ∼280 nm, probably due to the π–π* transition of the C–C bond. Furthermore, a single absorption peak appeared at ∼266 nm in the UV-vis spectra of rGO, indicating π–π* transitions of aromatic C–C bonds.14,22,23 However, the peak position and intensity of rGO and ACA-rGO differed significantly. It is interesting to note that the peak position of ACA-rGO was downshifted compared to that of the rGO. This may be attributed to the hindrance of the reduction process in the presence of ACA-rGO. The electron-donating properties may also contribute to the delocalization of the π-electron cloud of ACA with the rGO. The downshift in peak position was also an indication of the non-covalent interaction between the surface modifier and graphene.24
O bond. In contrast, after functionalization and reduction, the peak at 230 nm shifted to ∼256 nm, indicating the reduction of GO. Shoulder-like weak peaks appeared at ∼280 nm, probably due to the π–π* transition of the C–C bond. Furthermore, a single absorption peak appeared at ∼266 nm in the UV-vis spectra of rGO, indicating π–π* transitions of aromatic C–C bonds.14,22,23 However, the peak position and intensity of rGO and ACA-rGO differed significantly. It is interesting to note that the peak position of ACA-rGO was downshifted compared to that of the rGO. This may be attributed to the hindrance of the reduction process in the presence of ACA-rGO. The electron-donating properties may also contribute to the delocalization of the π-electron cloud of ACA with the rGO. The downshift in peak position was also an indication of the non-covalent interaction between the surface modifier and graphene.24
 |
| | Fig. 2 UV-vis spectra of GO, ACA-rGO and rGO. | |
3.3 FT-IR spectra analysis
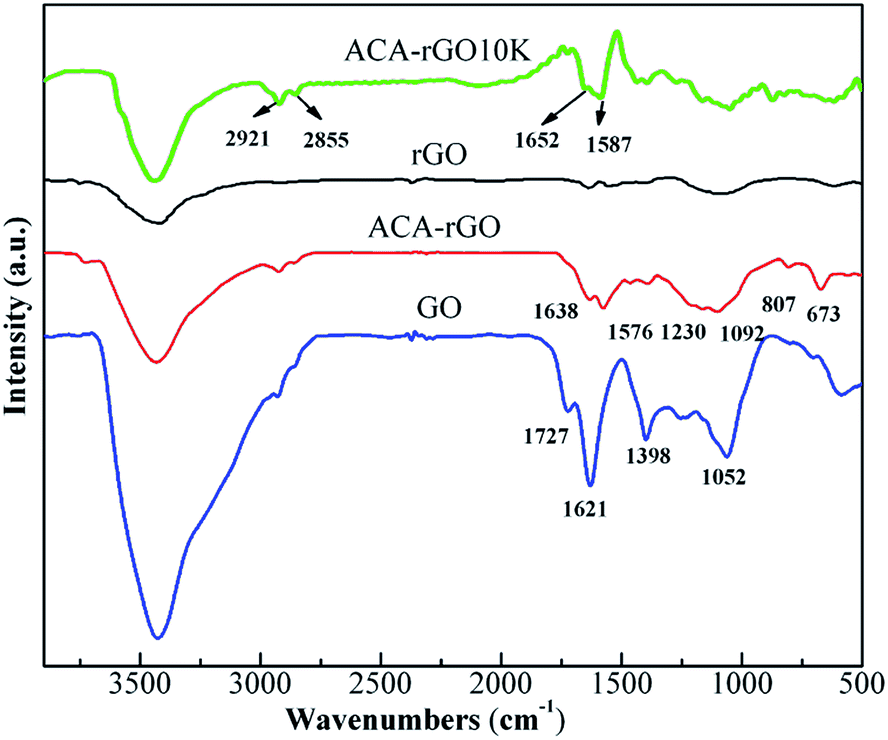
FT-IR spectra were carried out to identify the chemical nature of GO, rGO and ACA-rGO, as shown in Fig. 3. The FT-IR spectra of GO show high absorption peaks at 3423 and 1731 cm−1, indicating the presence of –OH and –COOH groups on the surface of GO during the oxidation of graphite. The highly intense peak of GO at 1621 cm−1 can be ascribed to the CC skeletal vibration of graphitic carbon. In addition, the absorption band from 1398 to 1052 cm−1 can be ascribed to the presence of oxygen moieties, such as hydroxyl, epoxy, and carbonyl groups. Interestingly, the peak intensities in the rGO decreased dramatically due to the removal of oxygen functionalities from the surface of the GO. In contrast, the intensities of the peaks in ACA-rGO were lower than that of GO but slightly higher than that of rGO, indicating a change in the chemical nature of ACA-rGO in the presence of the surface modifier. In ACA-rGO, the appearance of peaks at 3423 and 1638 cm−1 indicated the vibration of –OH on the surface of GO and CC skeletal vibrations of graphitic carbon, respectively.12,14,20 Furthermore, a new peak appeared in the ACA-rGO at 1576 cm−1 due to the aromatic ring of the modifier and the carbonyl and carboxylate groups.25 Therefore, FT-IR confirmed the presence of the modifier on the surface of the ACA-rGO. Peaks observed at 807 and 673 cm−1 corresponded to aromatic C–H stretching and out-of-plane bending vibration, respectively.
 |
| | Fig. 3 FT-IR spectra of rGO, ACA-rGO, GO and after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycle. 000 cycle. | |
3.4 Raman spectra analysis
Raman spectroscopy is an excellent tool for investigation of electronic structure and defect concentration in graphene. Raman spectra of GO, rGO, and ACA-rGO are shown in Fig. 4. The characteristic peak of graphitic materials (G-band at ∼1575 cm−1) was generated from the centre of the benzoic ring by the doubly degenerate phonon mode (E2g) of the sp2 carbon bond. In contrast, the D-band (1350 cm−1) was due to the breathing mode of k-point of A1g symmetry. In addition, the D-band was related with the vibrations of sp3 carbon atoms and electronic disorder in graphene. In pure crystalline graphite, the D-band is negligibly smaller than the G-band.26 The intensity of the D-band increased in GO, and the ID/IG ratio was 1.3. The blue shift of the G-band (1594 cm−1) in GO indicated the formation of isolated double bonds due to the extensive oxidation of graphite. In contrast, after reduction of GO, the ID/IG ratio decreased to 0.85, and the G-band shifted to 1583 cm−1, indicating the restoration of the π-electron cloud due to the solution phase reduction of GO. The D- and G-bands appeared at 1295 and 1591 cm−1, respectively, indicating the absorption of the ACA molecule on the surface of rGO. Interestingly, the 2D-band completely disappeared in the ACA-rGO. A similar phenomenon was observed in a previous report.27 The ID/IG ratio for ACA-rGO was 1.2, which can be ascribed to the absorption of ACA-anions on the surface of rGO. In addition, the blue shift of the G-band indicated a π–π stacking interaction between the ACA anion and the aromatic ring of grapheme.28 A comparative study of the Raman spectra indicated that the ACA anions were non-covalently attached to the surface of the rGO.
 |
| | Fig. 4 Raman spectra of GO, rGO and ACA-rGO. | |
3.5 XPS analysis
XPS was employed to analyse the chemical composition of ACA-rGO and rGO. The C1s spectra of ACA-rGO and rGO are depicted in Fig. 5(a) and (b). The curves were fitted using the Peak Fit EXEAISN software. XPS elemental analysis (Table 1) indicated that the oxygen moieties in rGO were lower than those of ACA-rGO. The higher concentration of oxygen functional groups in ACA-rGO was due to the existence of carboxylic acid functional groups of ACA and un-reacted oxygen moieties.
 |
| | Fig. 5 XPS C1s spectra of (a) ACA-rGO; (b) rGO. | |
Table 1 Elemental analysis of rGO and ACA-rGO
| Sample |
Carbon |
Oxygen |
Nitrogen |
O/C |
| rGO |
91.48 |
7.56 |
0.96 |
0.08 |
| ACA-rGO |
83.37 |
16.08 |
0.55 |
0.19 |
The presence of oxygen functional groups was confirmed by deconvolution of the C1s spectra of ACA-rGO and rGO. In ACA-rGO (Fig. 5(a)), the appearance of peaks at 284.5, 285.8, 287.6, and 288.3 eV reflected the presence of CC, C–O, CO/C–O, and CO bonds, respectively. Furthermore, the appearance of peaks at 289.5 and 290.2 eV indicated the presence of –COOH groups due to the ACA molecule and unreduced surface functional groups of GO. In addition, a less intense peak at 291 eV appeared which indicates that the sp2 character was maintained after reduction. In contrast, the C1s spectra of rGO (Fig. 5(b)) indicated the increased removal of oxygen functional groups from the surface of the GO.12,29–31
3.6 Thermogravimetric analysis
TGA of GO, rGO, and ACA-rGO was performed from room temperature to 700 °C at a heating rate of 5 °C min−1 under a N2 atmosphere, and the results are shown in Fig. 6. The thermal stability of GO was poor compared to that of the rGO and ACA-rGO due to the presence of labile oxygen moieties. The mass loss of GO up to 100 °C was ∼3%, indicating the absorption of moisture due to its highly hydrophilic nature. Significant mass loss of GO began in the temperature range of 160 to 270 °C due to removal of oxygen moieties from the surface of GO. The dramatic improvement in thermal stability of rGO was attributed to the removal of oxygen functional groups through hydrazine reduction. The final mass loss of rGO was ∼16%. In contrast, the thermal stability of ACA-rGO was higher than that of GO but lower than that of rGO. The initial mass loss of ACA-rGO was ∼3% (up to 164 °C) due to the removal of oxygen functional groups during reduction. Significant mass loss in ACA-rGO occurred in the region of 164–424 °C due to the pyrolysis of the remaining oxygen functional groups and ACA anions from the surface of ACA-rGO. Final mass loss was ∼28.4% at 700 °C, confirming the adsorption of ACA anions on the surface of rGO.14 UV-vis, FT-IR, Raman, and XPS analyses were in good agreement with the TGA measurements, further demonstrating the adsorption of ACA anions on the surface of rGO. Next, the electrochemical performance of ACA-rGO was investigated to examine to effects of ACA anions for supercapacitor applications.
 |
| | Fig. 6 TGA of rGO, ACA-rGO and GO. | |
3.7 Electrochemical performance
The capacitive performance of ACA-rGO, rGO, and GO were evaluated a using three-electrode system in which CV and galvanostatic charge–discharge were employed in a voltage window of −1 to 1 V. Comparative CV curves for ACA-rGO, rGO, and GO at a scan rate of 20 mV s−1 are shown in Fig. 7(a). The area covered by the ACA-rGO was higher than that of rGO and GO and was quasi-rectangular in shape. This quasi-rectangular CV curve had small redox peaks of ACA-rGO associated with the electrochemical double layer (EDL) rather than pseudocapacitance.32 In particular, the presence of the ACA anion increased the wettability with the electrolyte; further, removal of oxygen moieties from the surface of the grapheme increased the electron transition from the electrode to the electrolyte.
 |
| | Fig. 7 (a) CV curves of ACA-rGO, rGO, and GO at the scan rate 20 mVs−1 and CV curves of (b) ACA-rGO, (c) rGO, and (d) GO at scan rates of 10, 20, 50, 75, and 100 mV s−1. | |
The appearance of a redox peak indicated the electrochemical ion transition via the carboxylic anion of the ACA molecule and other oxygen moieties generated during oxidation of graphite. Fig. 7(b) shows the CV performance of ACA-rGO at different scan rate. It is apparent that, with an increased scan rate, the oxidation and reduction peak shifted right and left, respectively. These peak shifts with increasing scan rate indicate that ion transition was less prevalent than electron transition, and electrolyte ions were trapped in the interlayer of electrode materials.12 In contrast, the CV curves of rGO had similar rectangular nature; however, the area covered by the CV curves was less than that of ACA-rGO due to its hydrophobic nature. Further, the redox peak intensity was weaker than that of ACA-rGO, indicating fewer oxygen moieties than ACA-rGO, as indicated by XPS elemental analysis in Table 1. Moreover, the redox peak intensity of rGO decreased and was nearly parallel to the x-axis with increasing scan rate (Fig. 7(c)). In contrast, the redox peaks appeared in the ACA-rGO confirmed the presence of ACA anions on the surface of rGO. Furthermore, in GO, the CV curve (Fig. 7(d)) showed a high redox hump and an overall lower CV curve area. The lower curve area appeared due to the lower electrical conductivity of GO; however, the GO was highly dispersible in an aqueous medium. The presence of a large number of oxygen moieties on the surface of rGO increased the interactions within the aqueous medium but hindered electron conduction. Therefore, the CV curve area was smaller than that of ACA-rGO or rGO. Several redox peaks appeared in the CV curves, indicating ion exchange by several oxygen moieties, as implied by UV-vis spectra and FT-IR. The supercapacitor performance was examined using galvanometric charge–discharge curves.
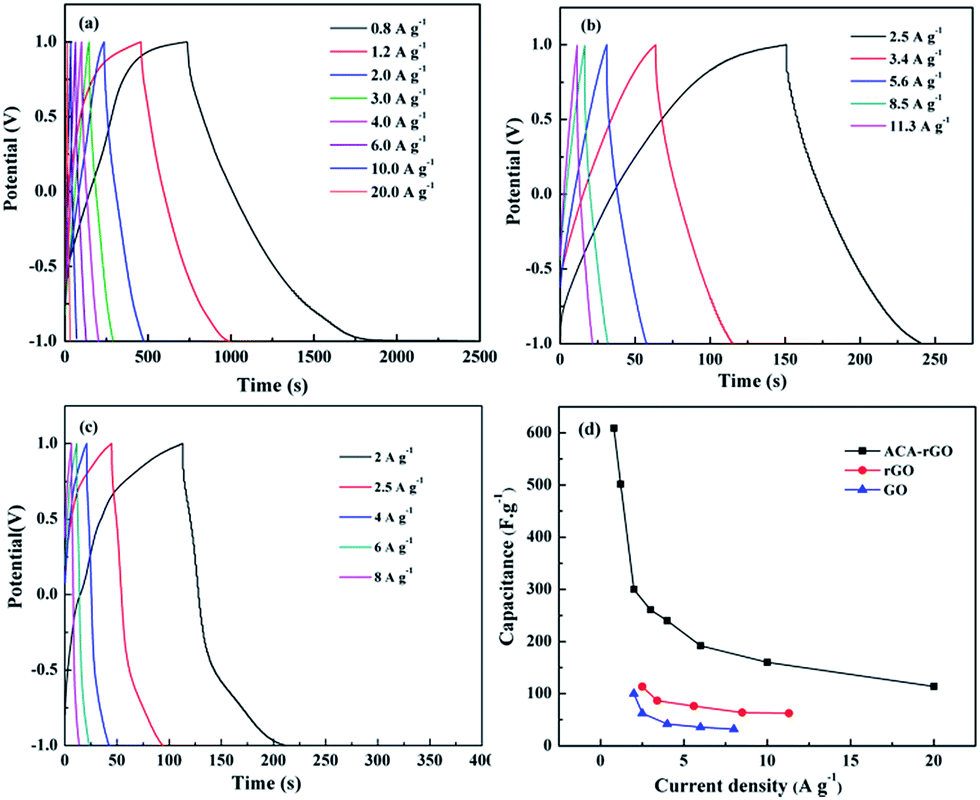
Fig. 8(a)–(c) show the charge–discharge curves with different current densities of ACA-rGO, rGO, and GO, respectively, which have a distorted rectangular shape due to a redox reaction. The capacitance of ACA-rGO was much greater than that of rGO or GO. The capacitance values calculated from the integration of CV curves were shown in Table S1 in the ESI.† The maximum capacitance values of ACA-rGO at the scan rate 10 and 100 mV s−1 were 425.8 and 271.6 F g−1, respectively. However, the capacitance values of rGO at the scan rate 10 and 100 mV s−1 were 218.4 and 110 F g−1, respectively. Moreover, the capacitance value of GO was quite lower than those of the ACA-rGO and rGO. The calculated capacitance value of GO was 144.4 and 36.2 F g−1 at same scan rates. The comparative galvanometric capacitance performance of ACA-rGO and rGO was shown in Fig. 8 at different current density. The shape of charge–discharge curve was not linear triangular in nature, indicating the capacitance performance was contributed by EDL and pseudocapacitance. The ACA anions of ACA-rGO are expected to participate in the redox reaction during the charge–discharge process and enhanced the total specific capacitance by contributing pseudocapacitance. Comparative capacitance values with different current densities are shown in Fig. 8(d). The range of specific capacitance values of ACA-rGO was 610 to 115 F g−1 at a current density of 0.8 to 20 A g−1. In contrast, the range of specific capacitance values of rGO was 102.3 to 62.3 F g−1 at current densities of 2.25 to 11.3 A g−1, while the range in GO was 100 to 32 F g−1 at current densities of 2 to 8 A g−1. The above results indicate that electrical conductivity and good wettability with the electrolyte are necessary to obtained equate capacitance performance.
 |
| | Fig. 8 Galvanometric charge–discharge curves of (a) ACA-rGO, (b) rGO, and (c) GO and (d) the specific capacitance values with different current densities of ACA-rGO, rGO and GO. | |
The capacitance performance of MWNTs was studied in three electrode system in Na2SO4 electrolyte to understand the electrode nature and the result is shown in Fig. S1 of the ESI.† The rectangular shaped CV curves at the voltage window of 0–1 V indicated that the capacitance performance was completely dominated by EDL nature. The highly mesoporous nature of MWNTs helped to transfer ions easily from electrode to electrolyte. In addition, the transfers of electron from MWNTS to the current collector during the electrochemical process were expected to be very fast due to the highly conducting nature of MWNTs. The specific capacitance was elucidated from the galvanometric charge–discharge graph (Fig. S2†) and highest specific capacitance of MWNTS was found to be 36.8 F g−1 at a current density of 0.4 A g−1. The conducting nature of MWNTS electrode was reflected in the EIS plot as shown in Fig. S3.† The capacitance performance of ACA-rGO in the range of −1–0 V was shown in Fig. S4 and S5.† The calculated specific capacitance of ACA-rGO in this range was 304 F g−1 at the current density 0.4 A g−1. These results indicated that ACA-rGO and MWNTS could be used as negative electrode and as positive electrode, respectively, for the fabrication of an asymmetric supercapacitor device to achieve better electrochemical performance.
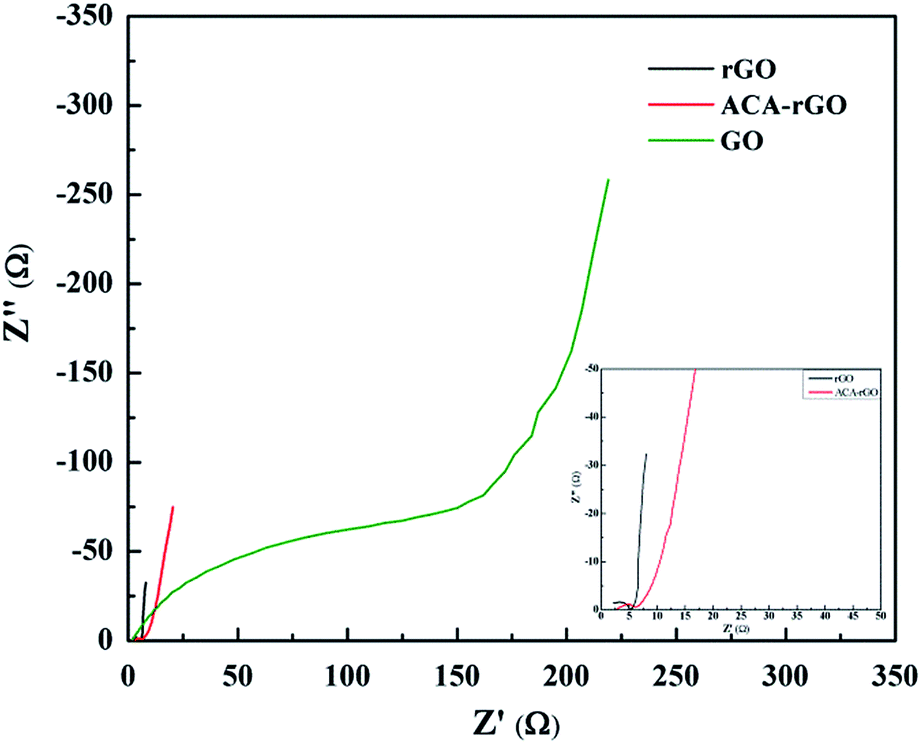
Electrochemical impedance spectroscopy (EIS) was performed to study the charge transfer process as a function of frequency at an open circuit voltage potential. Nyquist plots of rGO, ACA-rGO and GO are shown in Fig. 9. EIS of ACA-rGO showed a small semicircle in the high-frequency region and a linear slope in the low-frequency region in both rGO and ACA-rGO. The semicircles indicate the resistive behaviour and charge transfer process of rGO, ACA-rGO, and GO in Na2SO4 electrolyte in the high-frequency and mid-frequency regions, respectively. At high frequency, the resistance portion dominated and capacitance behaviour was negligible. Bulk solution resistance and charge transfer resistance were calculated from the EIS data. Bulk solution resistance was determined from the x-axis intercept at high frequency and was found to be ∼2.2 and 2.5 Ω for rGO and ACA-rGO, respectively. The charge transfer resistances were 5 and 6.4 Ω for rGO and ACA-rGO, respectively.33 The above results indicate that rGO and ACA-rGO are good electron conductors between the electrolytes and the electrode. In contrast, the EIS of GO clearly showed that the bulk solution resistance and charge transfer resistance were 3.8 and 250 Ω, respectively, indicating higher electron transfer resistance due to poor electrical conductivity.
 |
| | Fig. 9 Impedance spectra of rGO, ACA-rGO, and GO (inset the impedance spectra of rGO and ACA-rGO in short range). | |
The capacitance performance of ACA-rGO was studied using two-electrode configurations by asymmetric assembly with pure MWNTs. The two electrodes were separated by non-conducting porous PTEF film and packed into a sealed cell. The CV performance was studied at an operating voltage of −1 to 1 V. Fig. 10(a) shows the CV curve at various scan rates, which revealed that the curves were nearly rectangular in shape. However, at the terminating voltage at 1 V, a kink appeared, ascribed to decomposition of electrolyte and absorption of positive or negative ions by the electrode materials.32 Moreover, the rectangular nature of the CV curves suggests reversibility of the capacitive behaviour. The pseudocapacitance was contributed by the ACA anions of ACA-rGO and simultaneously EDL was dominated by MWNTS and rGO as well. Thus, the total capacitance (Ct) of the device was contributed by positive electrode capacitance (Cp) and negative electrode capacitance (Cn) and could be calculated as:
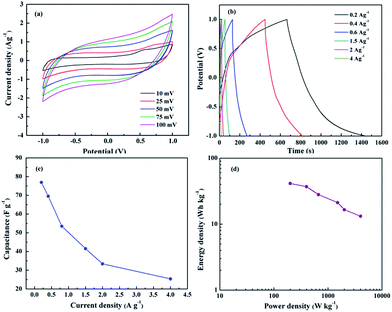 |
| | Fig. 10 (a) CV curves of asymmetric cell with ACA-rGO and CNT, (b) galvanometric charge–discharge curves, (c) cell capacitance value with different current density, and (d) Ragone plot of two electrode cell. | |
The values of Cp and Cn are same in a symmetric, but different in an asymmetric capacitors. In hybrid capacitor, the Ct value is closely related to the EDLC.34 In particular, when one of the electrodes in a hybrid supercapacitor is not polarisable or little polarisable, the potential of the other electrode is changing. Thus, the electrical capacitance can be doubled. The performance of hybrid capacitor has lower power characteristic than that of double layer capacitor due to the faradic reaction occurring in the cathodic terminal. Therefore, the Ct of pseudocapacitor/EDL hybrid capacitor is close to the double layer capacitor.35,36 The cell capacitance, energy density, and power density were evaluated from the galvanometric charge–discharge curves shown in Fig. 10(b). The calculated cell capacitance values were 77 and 25.3 F g−1 at current densities of 0.2 and 4 A g−1, respectively, as shown in Fig. 10(c). The highest energy density and power density were 41.36 Wh kg−1 and 4 kW kg−1, respectively, as shown in Fig. 10(d).
Impedance spectroscopy in Fig. 11(a) was performed to investigate the charge transfer process as a function of frequency. In two-electrode configurations, the equivalent series resistance (ESR) and charge transfer resistance values were found to be 0.68 and 6.38 Ω, respectively.33 Solvent resistance in two-electrode configurations was lower than that of the three-electrode system due to the close proximity of the anode and cathode. Impedance spectra indicated that the electron transition from electrolyte to nickel foam via ACA-rGO was minimal. In the low-frequency region, the two-electrode configuration showed a purely capacitive nature. The retention in cyclic stability of specific capacitance at a current density of 2 A g−1 was ∼80% after 10000 cycles as shown in Fig. 11(b). The above results indicated that ACA-rGO can be used as an electrode material for next-generation energy storage device.
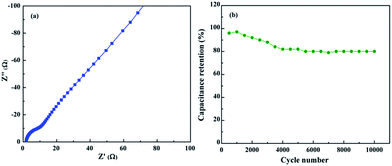 |
| | Fig. 11 (a) Impedance spectroscopy and (b) cyclic stability of the cell. | |
4. Conclusions
Herein, we successfully prepared functionalized aqueous dispersible graphene for supercapacitor applications. The morphological study indicated the formation of a few layers. Spectral analyses such as UV-vis, FT-IR, Raman, and XPS clearly indicated that the surface functional groups were changed in presence of ACA-anions. In addition, the thermal stability of ACA-rGO was significantly higher than that of GO but lower than that of rGO, indicating that the ACA anions were attached to the basal plane of reduced graphene by π–π interactions. Good electrochemical performance in a 1 M Na2SO4 aqueous solution was observed due to good wettability with ACA-rGO and an aqueous solvent. High electrochemical performance in a 1 M Na2SO4 aqueous solution was observed due to good wettability with ACA-rGO with aqueous solvent and showed higher capacitance value than rGO and GO. The cell capacitance values were 77 and 25.3 F g−1 at the current density 0.2 and 4 A g−1, respectively. The highest energy density and power density were 41.3 Wh kg−1 and 4 kW kg−1, respectively. The removal of oxygen from the surface and dipolar interactions between the ACA ions and aqueous solvent decreases the ESR value. Therefore, these materials will be applicable for future energy storage applications.
Acknowledgements
This study was supported by the Converging Research Center Program (2014M3C1A8048834) and the Basic Research Laboratory Program (2014R1A4A1008140) through the Ministry of Science, ICT & Future Planning and by the Basic Science Research Program through the National Research Foundation (NRF) funded by the Ministry of Education of Korea (2013R1A1A2011608).
Notes and references
- B. E. Conway, Electrochemical Supercapacitors: Scientific Fundamentals and Technological Applications, Kluwer Academic/Plenum Publishers, New York, 1999 Search PubMed.
- F. Béguin and E. Frackowiak, Carbons for Electrochemical Energy Storage and Conversion Systems, CRC Press, Taylor & Francis Group, 2010 Search PubMed.
- F. Stoeckli, A. Guillot, A. M. Slasli and D. Hugi-Cleary, Carbon, 2002, 40, 211–215 CrossRef CAS.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- C. Berger, Z. M. Song, X. B. Li, X. S. Wu, N. Brown, C. Naud, D. Mayo, T. B. Li, J. Hass, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. de Heer, Science, 2006, 312, 1191–1196 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- J. Xia, F. Chen, J. Li and N. Tao, Nat. Nanotechnol., 2009, 4, 505–509 CrossRef CAS PubMed.
- D. R. Cooper, B. D'Anjou, N. Ghattamaneni, B. Harack, M. Hilke, A. Horth, N. Majlis, M. Massicotte, L. Vandsburger, E. Whiteway and V. Yu, ISRN Condens. Matter Phys., 2012, 501686 Search PubMed.
- T. Kuilla, S. Bhadra, D. Yao, N. H. Kim, S. Bose and J. H. Lee, Prog. Polym. Sci., 2010, 35, 1350–1375 CrossRef CAS PubMed.
- T. Kuila, S. Bose, A. K. Mishra, P. Khanra, N. H. Kim and J. H. Lee, Prog. Mater. Sci., 2012, 57, 1061–1105 CrossRef CAS PubMed.
- P. Khanra, T. Kuila, S. H. Bae, N. H. Kim and J. H. Lee, J. Mater. Chem., 2012, 22, 24403 RSC.
- I. Momiji, C. Yoza and K. Matsui, J. Phys. Chem. B, 2000, 104, 1552–1555 CrossRef CAS.
- P. Khanra, T. Kuila, N. H. Kim, S. H. Bae, D. S. Yu and J. H. Lee, Chem. Eng. J., 2012, 183, 526–533 CrossRef CAS PubMed.
- R. B. Rakhi, W. Chen, D. Cha and H. N. Alshareef, Nano Lett., 2012, 12, 2559–2567 CrossRef CAS PubMed.
- Y. Cao and T. E. Mallouk, Chem. Mater., 2008, 20, 5260–5265 CrossRef CAS.
- D. Wang, P. Song, C. Liu, W. Wu and S. Fan, Nanotechnology, 2008, 19, 075609 CrossRef PubMed.
- E. Frackowiak and F. Béguin, Carbon, 2001, 39, 937–950 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, Preparation of graphitic oxide, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- J. Yan, Z. Fan, T. Wei, J. Cheng, B. Shao, K. Wang, L. Song and M. Zhang, J. Power Sources, 2009, 194, 1202–1207 CrossRef CAS PubMed.
- W. Chen, Z. Fan, L. Gu, X. Bao and C. Wang, Chem. Commun., 2010, 46, 3905–3907 RSC.
- T. Kuila, A. K. Mishra, P. Khanra, N. H. Kim, Md. E. Uddin and J. H. Lee, Langmuir, 2012, 28, 9825–9833 CrossRef CAS PubMed.
- Y. F. Jia, B. Xiao and K. M. Thomas, Langmuir, 2002, 18, 470–478 CrossRef CAS.
- C. Hu, Z. Chen, A. Shen, X. Shen, J. Li and S. Hu, Carbon, 2006, 44, 428–434 CrossRef CAS PubMed.
- K. Gao, Z. Shao, X. Wu, X. Wang, J. Li, Y. Zhang, W. Wang and F. Wang, Carbohydr. Polym., 2013, 97, 243–251 CrossRef CAS PubMed.
- H. M. A. Hassan, V. Abdelsayed, A. E. Rahman, S. Khder, K. M. AbouZeid, J. Terner, M. S. E. Shall, S. I. A. Resayes and A. A. E. Azhary, J. Mater. Chem., 2009, 19, 3832–3837 RSC.
- R. Voggu, B. Das, C. S. Rout and C. N. R. Rao, J. Phys.: Condens. Matter, 2008, 20, 472204 CrossRef.
- Z. Yang, X. Chen, C. Chen, W. Li, H. Zhang, L. Xu and B. Yi, Polym. Compos., 2007, 28, 36–41 CrossRef.
- V. Stengl, S. Bakardjieva, M. Bakardjiev, B. Stıbr and M. Kormunda, Carbon, 2014, 67, 336–343 CrossRef CAS PubMed.
- H. Kim, H. D. Lim, S. W. Kim, J. Hong, D. H. Seo, D. C. Kim, S. Jeon, S. Park and K. Kang, Sci. Rep., 2013, 3, 1–8 Search PubMed.
- M. J. Web, P. Palmgren, P. Pal, O. Karis and H. Grennberg, Carbon, 2011, 49, 3242–3249 CrossRef PubMed.
- W. Gu, M. Sevilla, A. Magasinski, A. B. Fuertes and G. Yushin, Energy Environ. Sci., 2013, 6, 2465–2476 CAS.
- M. N. Patel, X. Wang, D. A. Slanac, D. A. Ferrer, S. Dai, K. P. Johnston and K. J. Stevenson, J. Mater. Chem., 2012, 22, 3160–3169 RSC.
- W. G. Pell and B. E. Conway, J. Power Sources, 2004, 136, 334–345 CrossRef CAS PubMed.
- A. I. Beliakov and A. M. Brintsev, in Proceeding of the 7th International Seminar on Double Layer Capacitors and Similar Energy Storage Devices, Florida Educational Seminars, Dec 8–10, 1997 Search PubMed.
- J. H. Park, O. O. Park, K. H. Shin, C. S. Jin and J. H. Kim, Electrochem. Solid-State Lett., 2002, 5, H7–H10 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra12356e |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 , where I = instantaneous current, V1, V2 = cut-off potentials, and μ = potential scan rate, based on the cyclic voltammetry (CV) curve. Energy density (E) and power density (P) were calculated using the respective equations E = (1/2)CV2 and P = E/t.12,20,21
, where I = instantaneous current, V1, V2 = cut-off potentials, and μ = potential scan rate, based on the cyclic voltammetry (CV) curve. Energy density (E) and power density (P) were calculated using the respective equations E = (1/2)CV2 and P = E/t.12,20,21