
One-pot synthesis of UiO-66@SiO2 shell–core microspheres as stationary phase for high performance liquid chromatography†
Abstract
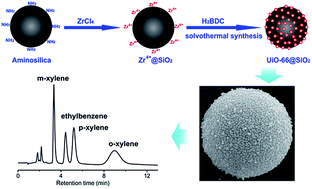
The unusual properties of ultrahigh surface area, adsorption affinity and shape selectivity make metal–organic frameworks (MOFs) a promising candidate as the stationary phase for high performance liquid chromatography (HPLC). However, the problems of high column backpressure and low column efficiency resulting from the direct packing of irregular MOF particles still remain in the HPLC separation. Herein, a facile one-pot synthesis method for the fabrication of MOFs@SiO2 shell–core microspheres was developed with aminosilica as the supporting substrate to grow the MOF shell. The density and particle size of the MOF shell could be easily controlled by adjusting the concentration of reactants, reaction temperature and time. UiO-66 (UiO for University of Oslo) was chosen as a model MOF because of its excellent chemical stability and unique reverse shape selectivity. The selected compounds including xylenes and ethylbenzene were effectively separated on the prepared UiO-66@SiO2 packed column with high resolution, good reproducibility and low column backpressure. The UiO-66@SiO2 packed column showed both reverse shape selectivity and a molecular sieving effect, making it attractive for the separation of structural isomers. Besides, the retention of analytes was also ascribed to the synergic effect of the hydrogen bonding between analytes and the amino groups of aminosilica, the hydrophobic effect and the π–π interaction between analytes and the aromatic rings of the UiO-66 shell.
 Please wait while we load your content...
Please wait while we load your content...

