
Asymmetric synthesis of (1R,2S)-1-amino-2-vinylcyclopropanecarboxylic acid by sequential SN2–SN2′ dialkylation of (R)-N-(benzyl)proline-derived glycine Schiff base Ni(ii) complex†
Abstract
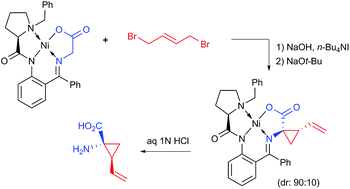
This work describes a new process for the asymmetric synthesis of (1R,2S)-1-amino-2-vinylcyclopropanecarboxylic acid of high pharmaceutical importance. The sequence of the reactions includes PTC alkylation (SN2), homogeneous SN2′ cyclization followed by disassembly of the resultant Ni(II) complex. All reactions are conducted under operationally convenient conditions and suitably scaled up to 6 g of the starting Ni(II) complex.
 Please wait while we load your content...
Please wait while we load your content...

