
Lipid/detergent mixed micelles as a tool for transferring antioxidant power from hydrophobic natural extracts into bio-deliverable liposome carriers: the case of lycopene rich oleoresins†
Disma Mastrogiacomoa,
Marcello Salvatore Lenuccia,
Valentina Bonfratea,
Marialuisa Di Caroloa,
Gabriella Piroa,
Ludovico Vallia,
Leonardo Resciob,
Francesco Milanoc,
Roberto Comparellic,
Vincenzo De Leocd and
Livia Giotta*a
aDipartimento di Scienze e Tecnologie Biologiche e Ambientali, Università del Salento, S.P. Lecce-Monteroni, I-73100 Lecce, Italy. E-mail: livia.giotta@unisalento.it
bPIERRE CHIMICA S.R.L., S.S. 476 Km 17, 650, I-73013 Galatina, LE, Italy
cCNR – Istituto per i Processi Chimico-Fisici, Sezione di Bari, via Orabona, 4, I–70126 Bari, Italy
dDipartimento di Chimica, Università di Bari, via Orabona 4, I-70126 Bari, Italy
First published on 2nd December 2014
Abstract
This work demonstrates that lipid-detergent mixed micelles can be employed successfully in order to achieve and modulate the transfer of bio-active hydrophobic compounds into lipid carriers by means of a simple and bio-safe procedure. In our specific investigation, liposome preparations incorporating mixtures of natural carotenoids with high lycopene content were developed and characterized, aiming to obtain formulations of potential nutraceutical and pharmaceutical interest. The starting material was a solvent-free high-quality lycopene rich oleoresin (LRO) obtained by extracting a freeze-dried tomato matrix with supercritical carbon dioxide (SC-CO2). Mixed micelles containing 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and cholate were loaded with LRO antioxidants by means of two slightly different procedures, which surprisingly resulted in significant differences in both quality and quantity of incorporated carotenoids. In particular, the selective incorporation of (all-E)-lycopene was achieved by extracting the oleoresin with a pre-formed cholate/POPC micelle suspension whilst (Z)-isomers were preferentially integrated when treating a POPC/LRO mixed film with cholate. The micelle to vesicle transition (MVT) method was employed in order to produce vesicles of well-defined lamellarity and size. Visible and infrared (IR) spectroscopy as well as Dynamic Light Scattering (DLS) and Transmission Electron Microscopy (TEM) measurements allowed the extensive characterization of LRO-loaded micelles and liposomes. The antioxidant potential of preparations was assessed by measuring the radical scavenging activity towards the coloured radical cation of 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonate) (ABTS). Important information about the reliability of different approaches for antioxidant capacity evaluation of micelle and liposome preparations was gained and the successful incorporation of LRO antioxidant power in a bio-deliverable water-dispersed form was demonstrated.
Introduction
Lycopene is a powerful antioxidant present in some fruits and vegetables1 and the most efficient quencher of singlet oxygen among carotenoids.2 Several studies have been devoted to the comprehension of lycopene effects on human health.3 This molecule has indeed shown anti-cancer,4 anti-inflammatory,5,6 and overall beneficial cardiovascular effects.7 However the uptake of lycopene from fruit and vegetable tissues is relatively poor because it is stored within chromoplasts as crystals or protein complexes which do not fully release the carotenoid during digestion in the gastrointestinal tract.8 Therefore, in order to exploit its health promoting properties, lycopene can be extracted from natural sources and used as nutraceutical ingredient in a suitable bio-available form. In this context, the extremely low solubility in water and consequently in biological fluids, as well as its chemical instability, require the use of appropriate carriers able to guarantee efficient therapeutics along with low administration doses.9 Liposomes are excellent candidates for these purposes, due to their biocompatibility, wide choice of physico-chemical properties and easy preparation. In spite of some drawbacks, which can arise from fair stability, poor batch-to-batch reproducibility, and low drug loading, liposomes offer indeed a number of advantages such as the possibility of encapsulating both hydrophobic and hydrophilic drugs, low toxicity and extreme versatility in the functionalization of their surface.10 Lipid vesicles have been extensively investigated as drug-delivery systems and recently proposed for increasing the nutritional bio-availability of vitamins in properly supplemented dairy products.11,12 Carotenoids incorporated in phospholipid bilayers have been extensively investigated13 not only aiming to develop nutraceutical preparations,14,15 but also as biomimetic model systems for elucidating the effects of these hydrophobic antioxidants on phase transition, fluidity, polarity and anisotropy of biomembranes,16,17 and for investigating the mechanisms of protection against lipid peroxidation.18,19 Some of these studies highlighted that, in spite of its extremely high antioxidant power, pure lycopene incorporated in lipid vesicles offers little protection against lipid peroxidation and shows a high degradation rate.18 Furthermore the synergistic effect of carotenoid mixtures in enhancing the antioxidant power has been widely demonstrated in biomembranes.20 These findings suggest that liposome preparations incorporating mixtures of natural carotenoids with a high lycopene content are expected to show enhanced efficacy than pure lycopene-loaded vesicles.A further aspect that can affect the biological activity of liposome-entrapped lycopene obtained from natural sources is the procedure adopted for carotenoid extraction from vegetables, since either undesirable contaminants or bioactive co-extracted compounds might worsen or improve respectively the therapeutic efficacy. Lenucci and co-workers have recently investigated agronomical, biological and technological aspects relevant to the preparation of a freeze-dried tomato matrix optimised for supercritical carbon dioxide (SC-CO2) extraction, producing a solvent-free high-quality lycopene rich oleoresin (LRO).21 In particular, the efficiency of the extraction method has been improved using a co-matrix constituted of roughly crushed hazelnuts, leading to a highly unsaturated vegetable oil where noteworthy amounts of lycopene, α-tocopherol, and phytosterols together with small but significant quantities of β-carotene and lutein are dissolved. Triglycerides with highly unsaturated acyl chains are the main constituents of the vegetable oil obtained, while phospholipids are completely absent.21
The aim of this work was to develop an effective procedure for incorporating the mixture of highly bio-active hydrophobic compounds of LRO in liposomes, thus increasing the water-solubility and bio-availability of lycopene and co-extracted antioxidants. The liposome formulation was settled using the micelle-to-vesicle transition (MVT) method,22 avoiding the introduction of any undesirable contaminant during the preparation, such as potentially toxic organic solvents, in order to preserve the original high bio-compatibility of the incorporated oleoresin. Compared to other liposome assembling techniques, the MVT method presents the advantage of producing homogeneous populations of unilamellar liposomes of controlled vesicle size,23 thus making easier the interpretation of subsequent studies relevant to their behaviour and action in biological systems. The size and size distribution of liposomes has indeed been demonstrated to strongly affect some important biological properties, such as organ/tissue selective bio-distribution24 and vesicle degradation rate in vivo,25 which in turn influence pharmacokinetics and pharmacodynamics of loaded drugs.26
Dealing with natural compound mixtures, synergistic effects and possible conversion in more or less active forms have to be considered in order to rationalize their bio-activity. Therefore the chemical composition of nutraceutical or pharmaceutical formulations based on natural compounds is often less important than their actual activity. Our effort was thus verifying the reliability of activity assays for micelle and liposome formulations in order to evaluate incorporation yields in terms of retained antioxidant activity rather than simply in terms of loaded chemical amount. The Trolox equivalent antioxidant capacity (TEAC) of LRO-loaded micelles and liposomes was estimated by means of the well-established assay based on the scavenging activity against the radical monocation ABTS˙+.27 This method has been applied to pure compounds and complex mixtures with different analytical strategies, namely decolourization assay, fixed-time point inhibition assay, reaction-rate-inhibition assay, and lag-phase measurement.28 We have employed two different approaches, by monitoring the antioxidant-induced attenuation of the chromogenic reaction producing ABTS˙+ by means of peroxyl radicals29 and by assessing the quenching power towards the preformed ABTS˙+ radical in the so-called decolourization assay.30 Attenuation and decolourization curves relevant to carotenoid-loaded vesicles and micelles were analysed aiming to elucidate both thermodynamic and kinetic properties of their radical scavenging power. Possible interferences arising from the presence of lipid components have been also investigated aiming to get useful information about the reliability of such antioxidant power assays to liposome formulations.
Experimental
Chemicals
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) was purchased from Lipoid GmbH, Ludwigshafen (Germany). Myoglobin from equine heart, hydrogen peroxide, 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS), and 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox) were purchased from Sigma. All other reagents and organic solvents employed were analytical grade and obtained from Sigma-Aldrich. Ultra-pure Milli-Q water was used for the preparation of any aqueous solution.Oleoresin preparation
The LRO was prepared following the procedure described in ref. 21. Briefly, 10 kg of tomato puree (cultivar Kalvert) was centrifuged at 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 10 min by using a J2-21 Beckmann centrifuge (Beckman Coulter, Fullerton, CA, US). The pellet was lyophilized by using a Christ ALPHA 2-4 LSC freeze-dryer (Martin Christ Gefriertrocknungsanlagen GmbH, Osterode am Harz, Germany). The freeze-dried tomato matrix was grinded at 500 μm by using a laboratory mill (Retsch GmbH, Haan, Germany) and blended (1:1 by weight) with roughly crushed hazelnuts (oleaginous co-matrix). The extraction was run using a laboratory scale apparatus (Spe-ed SFE system, Applied Separations, Allentown, PA, USA) fitted with a 25 mL stainless-steel extraction vessel (ø = 1 cm; h = 25 cm). About 25 g of the tomato–hazelnut mixture was packed into the vessel and dynamically extracted by flowing CO2 at a rate of ∼0.5 kg h−1, for 1 h. The other operative parameters were: pressure = 300 bar, and temperature = 60 °C. The oleoresin was stored at −20 °C until use for liposome preparation. The sample employed in the present study presented a lycopene content of 11.9 mg g−1 assayed by high pressure liquid chromatography (HPLC) analysis.
000 × g for 10 min by using a J2-21 Beckmann centrifuge (Beckman Coulter, Fullerton, CA, US). The pellet was lyophilized by using a Christ ALPHA 2-4 LSC freeze-dryer (Martin Christ Gefriertrocknungsanlagen GmbH, Osterode am Harz, Germany). The freeze-dried tomato matrix was grinded at 500 μm by using a laboratory mill (Retsch GmbH, Haan, Germany) and blended (1:1 by weight) with roughly crushed hazelnuts (oleaginous co-matrix). The extraction was run using a laboratory scale apparatus (Spe-ed SFE system, Applied Separations, Allentown, PA, USA) fitted with a 25 mL stainless-steel extraction vessel (ø = 1 cm; h = 25 cm). About 25 g of the tomato–hazelnut mixture was packed into the vessel and dynamically extracted by flowing CO2 at a rate of ∼0.5 kg h−1, for 1 h. The other operative parameters were: pressure = 300 bar, and temperature = 60 °C. The oleoresin was stored at −20 °C until use for liposome preparation. The sample employed in the present study presented a lycopene content of 11.9 mg g−1 assayed by high pressure liquid chromatography (HPLC) analysis.
Liposome preparation
LRO-incorporating liposomes were prepared using the MVT method as reported in ref. 31 with some modifications, that will be referred as method A. Briefly, an aliquot of LRO (2–20 mg) was weighted and mixed to a suitable volume of POPC ethanol solution (40 mg mL−1) in order to reach mass ratios LRO/POPC ranging from 0.1 to 10, roughly corresponding to lycopene/POPC molar ratios between 1:700 and 1:7 respectively. Added ethanol proved to solubilize only partially oleoresin-associated carotenoids, leading to a dark-reddish residue of undispersed material. The ethanol mixture was then dried by a gentle nitrogen flux to form a hydrophobic film on the walls of a conical glass tube. A 4% (w/v) sodium cholate solution containing 100 mM KCl and 50 mM K-phosphate (pH 7.0) was added to the dry lipid-oleoresin film in order to achieve a 1:10 phospholipid to detergent molar ratio. The final suspension thus contained cholate 4%, POPC 8 mg mL−1, and a variable amount of incorporated carotenoids depending on the LRO applied amount and the relevant carotenoid-incorporation yield. The detergent solution was sonicated for 10 min using a refrigerated ultrasonic bath operating at 35 kHz (Bandelin, Sonorex) to obtain the formation of mixed lipid/detergent micelles incorporating LRO. Undispersed light material was separated by centrifugation at 7000 × g for 3 min using 1.5 mL plastic microtubes and a bench-top microfuge (Hettich, Mikro 120). The resulting floating oil, represented mainly by oleoresin triglycerides, was removed and the remaining homogenous micellar dispersion was loaded on a glass chromatographic column (20 × 1 cm) packed with the gel filtration medium G-50 Sephadex (Sigma-Aldrich). The size exclusion chromatography was carried out using an elution buffer containing 100 mM KCl and 50 mM K-phosphate (pH 7.0). Liposomes eluted after a void volume of 1.5 mL. The elution profile was examined by collecting 200 μL fractions which were assayed for carotenoids, detergent and lipid concentrations.
A different protocol for obtaining LRO-loaded lipid/cholate micelles was also adopted (referred as method B), using a pre-formed micellar suspension containing 4% w/v sodium cholate and 8 mg mL−1 POPC in the same buffer (100 mM KCl, 50 mM K-phosphate, pH 7.0). 1 mL of this micellar suspension, showing the same POPC/detergent molar ratio (1:10), was mixed vigorously with a variable amount of LRO (2–20 mg) and analogously sonicated for 10 minutes. Afterwards the same steps described for method A were followed in order to obtain the relevant liposome preparation (Fig. 1).
 | ||
| Fig. 1 Main steps for the preparation of LRO-loaded POPC liposomes by methods A and B. Major differences in the (stereo)chemical composition of carotenoids loaded in micelles and liposomes are also schematized (see also Results and discussion section). | ||
UV-visible spectroscopy
Ultraviolet and visible spectra of solutions and suspensions were acquired by means of a Cary 5000 (Agilent Technologies) UV-Vis-NIR double beam spectrophotometer. The resolution was set to 1 nm, Hellma quartz cuvettes of 1 cm path length and suitable capacity were employed.Pigment extraction
Carotenoids in micellar and liposome dispersions were extracted with a 1:1:1 ethanol–n-hexane–diethyl-ether mixture as reported in ref. 18 and assayed by UV-visible spectroscopy. Briefly, 1 mL of aqueous sample was added to 2 mL of extraction mixture and mixed vigorously. After phase separation, a 1.1 mL of organic solution (upper phase), containing all carotenoids and hydrophobic components, was collected and analysed against a n-hexane blank. Since ethanol dissolves into the water phase upon mixing, the organic solvent which separates is represented mainly by a n-hexane–diethyl-ether mixture containing traces of water and ethanol, which favour the solubilisation of phospholipids into the organic phase. Spectra of LRO dissolved in this solvent and in n-hexane proved to present only negligible differences (data not shown) indicating that spectroscopic features of extracts can be compared directly to characteristics of standard carotenoids dissolved in n-hexane. The extraction yield of POPC by the mixture reported above was found to be around 90%, while pigment extraction yield was found very close to 100%.
Attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectroscopy
Infrared spectroscopy was employed in order to check cholate and lipid content in chromatographic fractions collected during the preparation of liposomes as reported in ref. 32. Mid-infrared spectra of dry films were acquired with a Spectrum One FTIR spectrometer (Perkin Elmer) equipped with a Deuterated Triglycine Sulfate (DTGS) detector and an ATR horizontal sampling apparatus. The internal reflection element (IRE) was a three bounce 4 mm diameter diamond microprism (Smith Detection, USA, former SensIR technologies). The spectral resolution used for all measurements was 4 cm−1. Typically, 16 interferograms were acquired and averaged for each spectrum. POPC concentration in chromatographic fractions was monitored following the signal at 1736 cm−1 (ester carbonyl stretching), while the band of the asymmetric carboxylate stretching vibration at 1555 cm−1 was used to follow cholate elution.Vesicle size assessment
Vesicle size of liposome preparations was assessed by their hydrodynamic radius obtained by DLS using the Horiba LB-550 Nanoparticle Size Analyzer (Horiba Jobin Yvonne). Measurements were performed at 25 °C with 100 sampling. The experimental-time-course changes in the measured light intensity were converted in the frequency distribution, or power spectrum (PS), by fast Fourier transformation. The particle size distribution was then obtained by comparing iteratively, using the Twomey method,33 the experimental PS with calculated frequency distribution based on the intensity of the Brownian movement for particles of different size.Transmission electron microscopy
Transmission Electron Microscopy (TEM) investigation of vesicles was performed with a JEOL JEM 1011 operated at 100 kV. Samples were prepared by casting a drop of a liposome suspension in MilliQ water onto carbon-coated copper grids. After 10 min, the excess suspension was removed and the wet grid was let to dry overnight. The films were negatively stained with a 2% (v/v) phosphotungstic acid solution for 30 s. The excess of phosphotungstic solution was removed by filter paper and the grid was let to dry overnight.Antioxidant activity assessment
In order to assess the antioxidant activity of as-prepared and vesicle-entrapped LRO, the kinetics of chromogenic reaction based on the production of the radical monocation ABTS˙+ was studied. H2O2 was employed as ABTS oxidizing species in presence of myoglobin as catalyst. For each assay myoglobin (4.5 μM final concentration) and ABTS (200 μM final concentration) were mixed in a 3 mL quartz cuvette filled with a 2.5 mL assay buffer containing 5 mM K-phosphate, 2 mM KCl, and 70 mM NaCl (pH 7). A time-course acquisition mode was selected, setting 734 nm as operating wavelength and 10 min as total running time. The initial reading of the spectrometer was zeroed and the signal was recorded for about 1 min under continuous stirring obtained with a cuvette stirring system (Rank Brothers Ltd, UK). The chromogenic reaction was then triggered by addition of 4.2 μL of a 30% H2O2 solution (150 μM final H2O2 concentration). A 30 μL aliquot of antioxidant standard or sample was added just before hydrogen peroxide addition. Experiments were run at room temperature. Temperature was checked by means of a thermocouple and its variability remained within 1 °C.The attenuation of the preformed ABTS˙+ radical in presence of LRO-containing liposomes and micelles was also investigated. For this purpose, ABTS was dissolved in phosphate buffer (5 mM K-phosphate, 10 mM KCl, and 60 mM NaCl, pH 7) to a 7 mM concentration and its radical cation was produced by addition of a sub-stoichiometric amount of potassium persulfate (2.45 mM final concentration), allowing the mixture to stand in the dark at room temperature for 12–16 h before use. ABTS oxidation occurs immediately, but the solution absorbance does not reach stable values until more than 6 h have elapsed. Afterwards the radical is adequately stable in presence of its unreacted neutral form for more than one month when stored in the dark at 10 °C. Nevertheless slow subsequent reactions involving the ABTS˙+ radical, such as disproportion processes, strictly required the employment of a double-beam set-up during measurements, in order to achieve a stable baseline. For this purpose an aliquot of radical solution, suitably diluted to reach a 0.7 absorbance value at 734 nm, was split into two 1 cm path length cuvettes (2 mL each) placed in the reference and sample holders of the spectrophotometer. After blank acquisition, 100 μL of Trolox standard or LRO-containing preparations were added to the sample cuvette and quickly mixed by means of the cuvette stirring system. Time course measurements demonstrated that decolourization for Trolox usually is complete within 1 min after mixing and subsequent absorbance drifts are negligible for at least 20 minutes. Since for micelle and liposome samples decolourization was slower, absorbance readings were taken 6 min after antioxidant addition. The resulting negative ΔA values (At=6 min − At=0) and the absorbance of the initial carbocation solution at the same wavelength (A0) were used for calculating the ABTS˙+ decolourization ratio (−ΔA/A0). The calibration curve was built using Trolox as hydrosoluble antioxidant standard in order to obtain the TEAC of samples. All determinations were carried out in triplicate at each different concentration of the standard and samples.
The same assay was modified in order to analyse hydrophobic oleoresin samples. In this case a 100 μL aliquot of a n-hexane solution of the sample or standard was added to 1 mL of the ABTS˙+ aqueous solution and shaken vigorously for 2 min. Afterwards the organic phase was separated by centrifugation (7000 × g for 2 min) and pipetted away. The decolourization extent, due to ABTS˙+ neutralization, was assayed analysing the aqueous phase as described above. The blank was prepared for each sample in parallel using 100 μL of pure n-hexane. The hydrophobic compound α-tocopherol was used as standard antioxidant. The antioxidant capacity (referred as α-TEAC) was thus given in α-tocopherol equivalent units.
Results and discussion
LRO UV-visible spectra
As mentioned above the technique employed for extracting lycopene from a freeze-dried tomato matrix leads to a complex mixture whose the basic component is hazelnut oil. This major component is mainly constituted by triglycerides that do not absorb in the visible range, as well as α-tocopherol and phytosterols. Thus the pigments responsible for the red-orange colour of the preparation are exclusively carotenoids (∼1%) of which lycopene is the most abundant (>97%).21 In general the visible absorption spectrum of carotenoids is strongly influenced by the solvent and by the relative amounts of stereoisomers in the sample. (all-E)-forms, possessing the extended coplanar structure, present the greatest colour intensity, while (Z)-isomers show decreased intensities and lower wavelength of the absorption maxima.34 The absorption spectrum in the 250–650 nm range of LRO dissolved in n-hexane, shown in the left panel of Fig. 2 (dotted line), is consistent with a significant amount of lycopene (Z)-isomers in the preparation, since the expected maxima of (all-E)-lycopene in n-hexane at 444, 471 and 502.5 nm (ref. 35) are shifted to lower wavelength values. | ||
| Fig. 2 Left panel: UV-visible spectra of LRO dissolved in n-hexane (dotted line) at the concentration of 0.4 mg mL−1, and of pigments extracted from LRO-loaded micelles prepared by method A (solid thin line) and method B (solid bold line); the control spectrum (dashed line) is relevant to the solution obtained extracting plain cholate/POPC mixed micelles and represents the contribution of components other than LRO pigments. Right panel: UV-visible spectra of LRO-loaded micelle suspensions prepared by method A (thin line) and method B (bold line) and of a plain micelle suspension (dashed line). The LRO concentration applied for loading the micelles was 20 mg mL−1. Dilution factor of both extracts and micelles was 5. | ||
Moreover bands around 350 nm, absent in (all-E)-lycopene and β-carotene spectra, are characteristic of (Z)-isomers of lycopene.34 β-Carotene, with characteristic absorption maxima at 452 and 479 nm, likely contributes to broaden the absorption bands of LRO at higher wavelength values.
LRO-loaded mixed micelles
The right panel in Fig. 2 shows the UV-visible spectrum of a POPC/cholate micelle suspension incorporating LRO obtained following the method A (thin line). Hydrophobic components were extracted as described in the Experimental section and the spectrum of the resulting organic solution is reported in the left panel (thin line). The comparison with LRO spectrum reveals that micelle-incorporated carotenoids do not have the same (stereo)chemical composition of original LRO. In particular the band characteristic of LRO at 498 nm appears here only as a weak shoulder indicating an even lower relative concentration of (all-E)-lycopene in the micelle extract. Moreover the analysis of oil phase recovered after the centrifugation step shows that LRO pigments not incorporated in micelles present a higher (all-E)-lycopene relative content, i.e. higher absorption at 502.5 nm, supporting the hypothesis that a selective uptake of carotenoid and lycopene isomers occurs during the incorporation process.The change of carotenoid composition following the micelle integration process prevents any accurate spectroscopic quantification of LRO incorporation yield. However a qualitative analysis of spectra revealed that isomeric composition of lycopene in micelles is affected likewise by the LRO applied amount. In particular the increase of applied LRO mass enhances both the overall amount of loaded pigments and the (Z) to (all-E) molar ratio of loaded lycopene (see ESI†).
Fig. 2 shows also analogue spectra relevant to an LRO-loaded micellar system prepared following method B (right panel, bold line). In this case the oleoresin was let to interact with a pre-formed POPC/cholate mixed micelle suspension. Interestingly this slight modification produced significant changes in the final preparation. The spectrum of the extract (left panel, bold line) presents indeed neat peaks at 471 and 503 nm characteristic of (all-E)-lycopene, indicating that this isomer is widely predominant in micelles. Furthermore, as can be seen from the comparison between extract and micelle spectra, the micelle environment proved to strongly modify the spectral features of incorporated (all-E)-lycopene resulting in wide hypsochromic shifts and significant hypochromic effect.
Since the selective incorporation of a specific stereoisomer allows its accurate quantification by visible spectroscopy, the dependence of micelle-incorporated amount of (all-E)-lycopene from the applied LRO amount was investigated. Fig. 3 shows that it is possible to enhance significantly micelle-loaded amount by increasing the LRO mass applied to the POPC/cholate pre-formed micellar system.
 | ||
| Fig. 3 The concentration of (all-E)-lycopene transferred in the water phase by incorporation in mixed micelles as a function of LRO mass applied to the pre-formed POPC/cholate micellar suspension. The upper X axis indicates the ratio between LRO and POPC masses in the starting oil–water biphasic system. | ||
On the basis of these results it is evident that the amount of carotenoids in POPC–cholate mixed micelles can be enhanced by increasing the applied LRO mass for both the adopted procedures. Nevertheless, the specific method employed for micelle preparation strongly affects the final composition of loaded carotenoids leading to almost complementary results: using a pre-formed POPC–cholate mixed micelle suspension the selective incorporation of (all-E)-lycopene is achieved, whilst a preferential incorporation of (Z)-lycopene isomers and other carotenoids is obtained when a cholate solution was allowed to interact with a pre-formed LRO/POPC film (see also Fig. 1).
In the latter case the limited incorporation of (all-E)-lycopene in micelles is likely linked to the low solubility of this isomer in ethanol, which in turn does not allow its effective dispersion in the POPC layer, subsequently interacting with cholate. When the POPC ethanol solution is added to LRO, a dark reddish undispersed material actually separates, which presented spectroscopic features of (all-E)-lycopene. The following ethanol evaporation likely results in an inhomogeneous hydrophobic layer where a phase represented by LRO well dispersed in POPC coexists with small (all-E)-lycopene crystals.36 Subsequently, only the phase containing POPC is able to effectively interact with the cholate solution leading to the formation of mixed micelles.
The major role played by POPC in promoting LRO incorporation in micelles was further demonstrated by analysing the visible spectrum of a 4% cholate solution sonicated for 2 min with LRO and successively centrifuged for separating undispersed material. The spectrum of the resulting colourless aqueous phase did not show any signal typical of carotenoid pigments, demonstrating that the small cholate micelles are not able to effectively disperse large carotenoid molecules.
Micelle preparations were further characterized by infrared spectroscopy, aiming to get information on the incorporation of components other than pigment molecules. The ATR-FTIR spectra of dry films obtained by casting/evaporation technique from plain and LRO-loaded POPC/cholate micelles prepared by method A are reported in Fig. 4 (traces D and E respectively). Spectra present high similarity, demonstrating that loaded material amount is well below the bulk POPC/cholate amount. The infrared spectrum of micelles prepared by method B is analogue to trace D as well (data not shown). Spectra of cholate (trace A) and POPC (trace B) cast films are also reported in the same figure for comparison. Asymmetric and symmetric COO− stretching bands at around 1560 and 1400 cm−1 respectively, arising from cholate carboxylate groups, are well visible in micelle spectra together with the absorption band of ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1736 cm−1. This signal appears as a shoulder due to superimposition of water bending band at 1650 cm−1 arising from the presence of hydrated charged functional groups such as phosphate and carboxylate. The mid-infrared spectrum of LRO deposited on the ATR crystal (trace C) presents an intense absorption band at 1740 cm−1 due to vibration of CO ester bond of glycerides and a characteristic weaker band at 1653 cm−1 ascribable to their highly unsaturated acylic chains (CC stretching mode). The incorporation of LRO glycerides would then result in enhancement of ester CO signal. Since the intensity ratio between bands at 1736 cm−1 and 1560 cm−1 in plain and loaded micelles appears instead the same, the incorporation of LRO glycerides can be considered negligible for both methods A and B.
O stretching at 1736 cm−1. This signal appears as a shoulder due to superimposition of water bending band at 1650 cm−1 arising from the presence of hydrated charged functional groups such as phosphate and carboxylate. The mid-infrared spectrum of LRO deposited on the ATR crystal (trace C) presents an intense absorption band at 1740 cm−1 due to vibration of CO ester bond of glycerides and a characteristic weaker band at 1653 cm−1 ascribable to their highly unsaturated acylic chains (CC stretching mode). The incorporation of LRO glycerides would then result in enhancement of ester CO signal. Since the intensity ratio between bands at 1736 cm−1 and 1560 cm−1 in plain and loaded micelles appears instead the same, the incorporation of LRO glycerides can be considered negligible for both methods A and B.
 | ||
| Fig. 4 ATR-FTIR spectra of sodium cholate (trace A), POPC (trace B), LRO (trace C), plain POPC/cholate micelles (trace D), and LRO-loaded POPC/cholate micelles prepared by method A (trace E). Dry films onto the ATR crystal were obtained by casting (sample C) or by casting/evaporation technique using aqueous (samples A, D, and E) or ethanol (sample B) solutions. | ||
Liposome production and characterization
Micelle preparations obtained by both methods were loaded on Sephadex G-50 Medium packed chromatographic columns and the relevant elution profiles were analysed. Fig. 5 shows a typical elution profile obtained by means of spectroscopic measurements in the infrared and visible range. 2 μL of liposome suspension were cast and dried on the ATR prism for recording the infrared absorption due to POPC and cholate, whilst visible absorption of the suspension was recorded using the conventional transmission mode. In the latter case scattered light contribution was suitably subtracted. Since carotenoids and POPC eluted simultaneously, the effective association of pigments and lipids in supramolecular structures was demonstrated. However, the detergent depletion during the chromatographic run was not complete in our experimental conditions since elution bands of lipids and cholate did not appear perfectly resolved. Detergent-free lipid fractions represented typically the 60–65% of applied lipid mass. The remaining lipid amount, eluting with a minimal detergent contamination, was discarded since the lipid vesicular organization was not assured in these fractions. In spite of the low MVT yield due to low column resolution, this detergent-depletion method proved to be extremely fast (around 2 minutes from micelle loading to liposome collection) and was therefore adopted. | ||
| Fig. 5 The typical elution profile obtained applying 0.5 mL of LRO-loaded POPC–cholate micelles on a gel-filtration (Sephadex G-50 Medium) packed chromatographic column. See Fig. 4 for POPC and cholate marker bands. | ||
The rapidity of the procedure is indeed of noteworthy importance when dealing with the incorporation of highly unstable molecules such as antioxidants. The analysis of collected fractions revealed that the loss of lipids and carotenoids in the final detergent-free suspension was higher in the case of micelles prepared by method B, indicating that the characteristics of loaded micelle sample affects the effectiveness of the detergent depletion process during the chromatographic run.
Fig. 6 shows the visible spectra of liposome preparations and of the relevant pigment extracts obtained by both methods starting from the same LRO mass (20 mg).
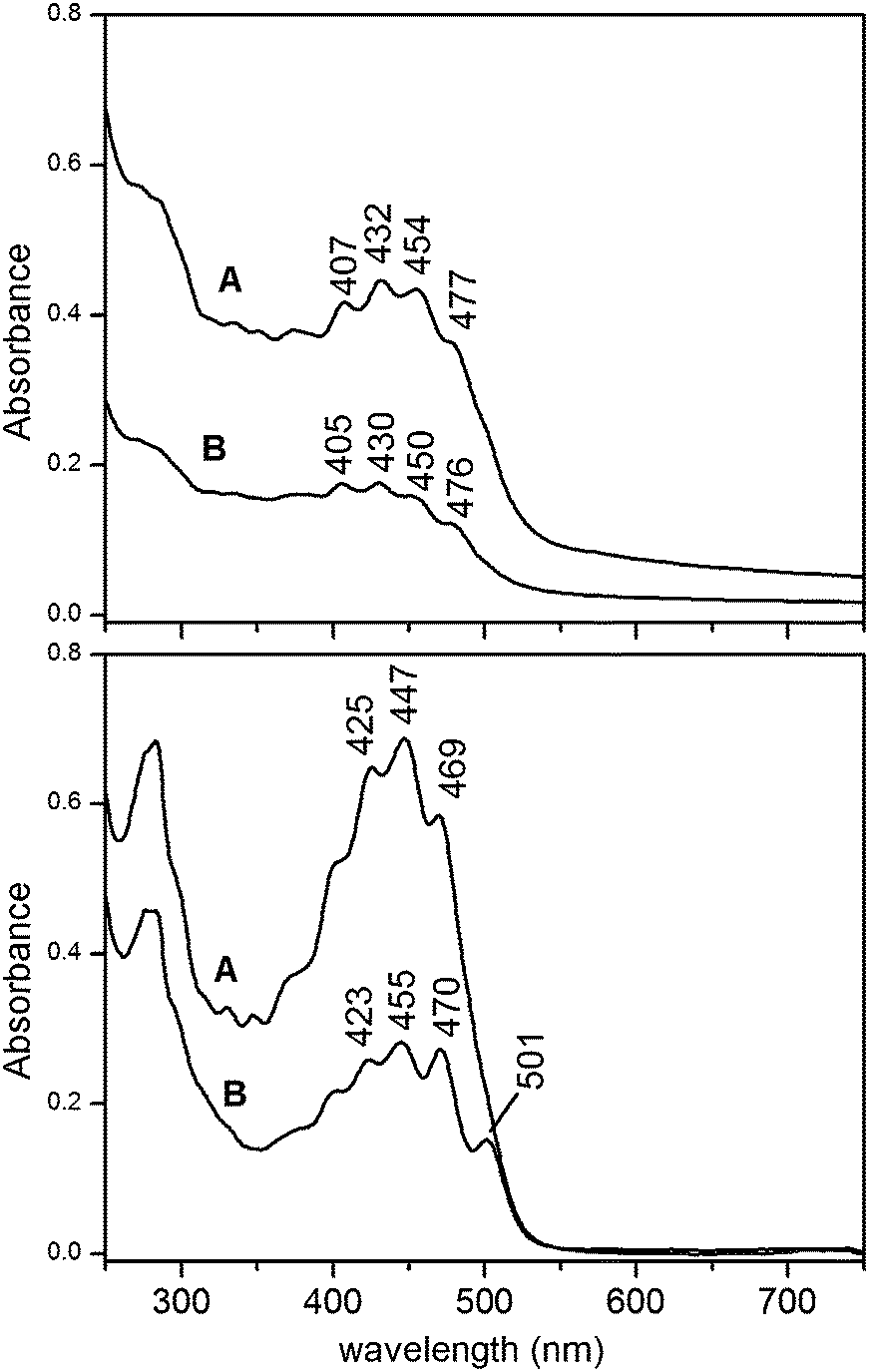 | ||
| Fig. 6 Visible spectra of as prepared LRO-loaded vesicle suspensions obtained adopting method A and B (upper panel). 20 mg of LRO per mL of detergent solution (method A) or per mL of mixed micelle dispersion (method B) were employed. The lower panel shows the spectra of pigments extracted from the relevant LRO-loaded-liposomes. | ||
The comparison of signal intensities in spectra relevant to method A and B reveals that the former allows getting more concentrated carotenoid preparations. Moreover, the stereo-isomeric composition of incorporated lycopene is quite different in the preparations following the selective uptake described for the micelle incorporation process.
DLS measurements revealed that liposomes prepared by both methods present a hydrodynamic diameter of around 40 nm. The relevant size distribution for both preparations is presented in Fig. 7. Repeated DLS measurements allowed verifying that size distribution remains unchanged even after four days at room temperature, thus demonstrating the good physico-chemical stability of preparations.
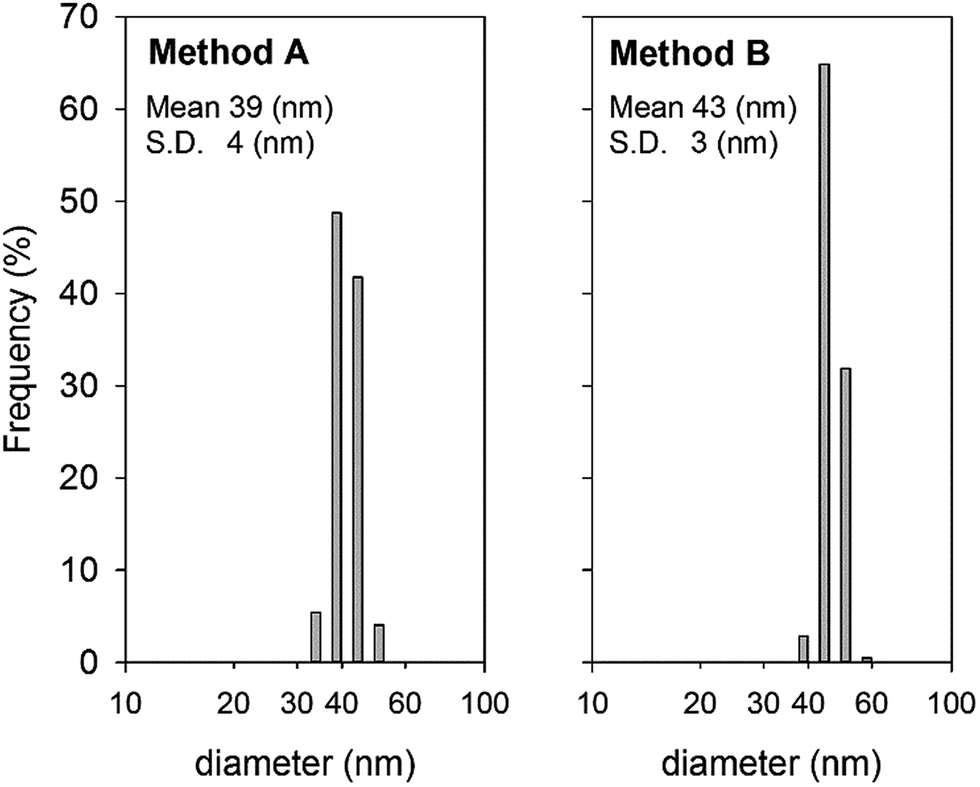 | ||
| Fig. 7 Hydrodynamic diameter distribution of liposomes prepared by methods A and B, obtained by dynamic light scattering. Data were recorded at 25 °C. | ||
The vesicular structure of lipid carriers obtained by our protocol was confirmed by TEM analysis. Fig. 8 shows that the MVT method was unambiguously able to generate small vesicles, whose mean diameter was consistent with DLS data. TEM measurements do not allow to demonstrate unilamellarity of our vesicles. Nevertheless it is well known that the MVT method employing a fast detergent removal process produces unilamellar monodisperse vesicles.22 Moreover the unilamellarity of vesicles analogously prepared by size exclusion chromatography, as fast detergent depletion method, has been recently pointed out by de Leo and coworkers by means of Cryo-EM measurements.37 Therefore LRO-loaded vesicles described in this work can be safely considered unilamellar.
 | ||
| Fig. 8 TEM image of LRO-loaded liposomes prepared by method A. | ||
The small size of the unilamellar vesicles prepared by our protocol accounts for the minimal turbidity of their suspensions, as can be seen in Fig. 6 (upper panel), which makes them suitable for optical studies addressed to elucidate their activity. Moreover, the well-defined size and lamellarity characteristics are interesting in order to investigate their ability to interact with cellular systems.
Antioxidant activity assessment
The antioxidant capacity of liposomal preparations was assessed spectrophotometrically by investigating their influence on the rate of the reaction leading to the formation of the coloured ABTS˙+ radical (showing maxima at 415 and 734 nm). In the presence of antioxidants such as Trolox, the chromogenic reaction, catalysed by myoglobin and initiated by hydrogen peroxide, has a lag period, and its duration is linearly correlated to the concentration of the antioxidant. The lag period indicates that the antioxidant is able to react with the oxidized intermediate of myoglobin (ferrylmyoglobin), thus preventing ABTS˙+ formation and accumulation. Alternatively the lag-time phenomenon may exist in presence of a very fast ABTS˙+ neutralization rate.Fig. 9 depicts the kinetic curves obtained with the standard Trolox and LRO-loaded liposomes in comparison with the control conditions (no antioxidant added). LRO-loaded liposomes clearly do not show a Trolox-like antioxidant mechanism, since sample addition does not induce any lag-time appearance. LRO-loaded liposomes are instead able to lower the ABTS˙+ production rate. A similar behaviour was reported by Yu et al.29 for flavonoids apigenin and genistein whose antioxidant capacity acts via a slow-rate ABTS˙+ quenching, and for Fe-chelating agents, such as EDTA, which obstruct only partially the regeneration of myoglobin Fe(IV).
 | ||
| Fig. 9 Kinetics of myoglobin-catalysed oxidation of ABTS by H2O2. The chromogenic reaction, producing the coloured radical ABTS˙+, is delayed in presence of Trolox, whilst a lag time does not appear in the case of LRO-loaded liposome addition. | ||
Some commercial antioxidant assay kits adapted the lag-time method in order to run faster TEAC measurements, without the needing of recording kinetic curves for both sample and standards. In this case, a stop solution, which denatures the myoglobin catalyser thus instantly blocking the reaction, is employed. The ABTS˙+ amount at the end point is inversely dependent on the extent of the lag time, so that a calibration curve can be built plotting single absorbance readings at the end point as a function of standard concentration. However such an approach can be utilized only for antioxidants with a fast reaction rate with ferrylmyoglobin or which can quench ABTS˙+ rapidly. In fact, the fast consumption of the antioxidant during the lag period allows the reaction, once started, to proceed with the same rate than control. The application of this method to antioxidants reacting slowly with the chromogenic radical would provide inaccurate values strongly dependent on the chosen stopping time. Therefore, a preliminary kinetic curve analysis should always be carried out in order to evaluate the method reliability, even though this issue is not always clearly stated in the commercial kit instructions.
In our case kinetic curve analysis reveals that the myoglobin/ABTS method is not reliable. The graph in Fig. 9 clearly shows that TEAC value of LRO-loaded liposomes provided by the assay would even double increasing the stopping time from 3.5 to 4.5 minutes.
Moreover, the presence of phospholipids in our samples further complicates the interpretation of data arising from ABTS chromogenic reaction kinetic curves. Phospholipids alone (tested using plain liposome preparations) proved to slow down the ABTS˙+ production rate in the myoglobin-catalysed reaction (data not shown), even though subsequent measurements revealed that they are not able to decolourize pre-formed ABTS˙+ solutions. This finding demonstrates that phospholipids interfere with the enzyme reaction, likely competing with ABTS for oxidation. Myoglobin-induced lipid oxidation is indeed a well-known phenomenon in biological systems.38 In addition, micelle samples cannot be analysed by this assay, since the detergent affects integrity and activity of the myoglobin catalyser.
The reliability of the ABTS˙+ decolourization assay was then assessed. Micelle and liposome samples were added to a preformed ABTS˙+ radical solution and the ability to scavenge this radical was evaluated. Decolourization curves are presented in Fig. 10. Negative ΔA values account for the absorbance decrease at 734 nm following the conversion of the radical ABTS˙+ into the neutral colourless form. The quenching of the pre-formed radical by the standard antioxidant Trolox was found very fast (trace B), in agreement with its behaviour in delaying myoglobin-catalysed reaction. A slight recovery of the absorbance was observed after the initial bleaching, leading however to stable readings after 1 min from sample addition. In order to assess the role of mixed micelle environment in affecting the ABTS˙+ quenching ability, the complete incorporation of α-tocopherol was achieved in POPC/cholate micelles by the standard MVT protocol,31 and the relevant decolourization curves were analysed. Micelle-associated α-tocopherol produced the same absorbance drop than an equivalent concentration of its soluble derivative Trolox, showing an analogue kinetic behaviour (trace A). This finding demonstrates that either POPC or cholate do not interfere with the reaction between ABTS˙+ and the reactive moiety of α-tocopherol molecule. The antioxidant power of aqueous samples analysed by the decolourization assay can be therefore referred indistinctly as TEAC or α-TEAC, letting a better comparison with hydrophobic samples such as the oleoresin. The antioxidant capacity of LRO was indeed determined (α-TEAC = 17.4 μeq g−1) by a modified decolourization assay employing a biphasic water–n-hexane system and α-tocopherol as standard compound (see Experimental section). By means of this assay, the carotenoid-free oleoresin, obtained by extracting a tomato-free hazelnut matrix, was also analysed, revealing that the contribution of the hazelnut oil components to the overall antioxidant power of LRO is minor (α-TEAC = 0.3 μeq g−1).
 | ||
| Fig. 10 Decolourization curves obtained adding to a stirred ABTS˙+ solution the following antioxidants: α-tocopherol 48 μM incorporated in POPC/cholate mixed micelles (A), Trolox 72 μM (B), plain POPC liposomes (C), LRO-loaded POPC liposomes prepared by method B (D), LRO-loaded POPC liposomes prepared by method A (E), LRO-loaded POPC/cholate mixed micelles prepared by method B (F), LRO-loaded POPC/cholate mixed micelles prepared by method A (G). | ||
The right panel in Fig. 10 shows the decolourization curves relevant to different liposome and micelle samples. The ability of plain POPC liposomes to quench ABTS˙+ was found negligible (trace C). This finding is in agreement with measurements carried out on α-tocopherol-loaded micelles described above, which excluded any ABTS˙+ quenching activity by mixed micelle components, such as POPC.
Since LRO-loaded liposomes and micelles were analysed as prepared, the extent of the decolourization was strongly influenced by both their carotenoid content and their concentration. The higher carotenoid content accounts for wider ΔA values arising from preparations obtained with method A (traces E and G versus D and F), whilst the sample dilution and the loss of material following the chromatographic run results in a significant decrease of the decolourization ability of liposomes with respect to micelles (traces F and G versus D and E).
The decolourization curves recorded with liposome and micelle samples showed an initial rapid absorbance drop, followed by a slower absorbance decay. A similar behaviour was reported for several antioxidants, such as resorcinol,39 rutin,40 chrysin41 and other flavonoids,42 and smartly explained with the formation of reaction products acting in turn as ABTS˙+ scavengers.41 Hence secondary reaction products of carotenoids dispersed in our lipid matrices give likely a considerable contribution to TEAC, acting as ABTS˙+ neutralizers with a kinetics slower than that one of the parent compounds.
Decolourization curves for complex antioxidants mixtures like wines, reported by Villano et al.,43 are also analogue to those obtained for our carotenoid-containing samples, indicating a weak but significant time dependence of the TEAC values determined by this assay. The same authors assigned to wines a “fast” scavenging activity relevant to the decolourization achieved in the first 2 min and a “total” scavenging activity obtained reading the absorbance drop at the end point of 15 min. On the other hand, TEAC at 10 s was referred as “fast” by other authors41,42 and the “total” TEAC was often fixed at 6 min.41 In agreement with the latter practice, we assigned total TEAC values to LRO-loaded liposomes and micelles reading ΔA values 6 min after sample addition. Although a plateau ΔA value was not reached at this time, the resulting underestimation of the overall antioxidant capacity appeared however acceptable. We preferred to follow the “total” TEAC value, rather than the “fast”, during the incorporation process, since this value was found to better correlate with the inhibition of lipid peroxidation.42 Moreover it should be mentioned that ABTS˙+ has a relatively slow reactivity as compared to physiological important radicals, so that the slow scavenging reaction of secondary products might be much faster in vivo and play an important role in protection mechanisms.42
Any absorbance drift following the initial decolourization was not observed in the case of the modified assay employed for α-TEAC evaluation of LRO and hazelnut oil (see ESI†). In this procedure the removal of the n-hexane phase containing the antioxidant after a 4 min contacting time with the ABTS˙+ solution allows stopping any slow reaction leading to stable absorbance values. However, to rule out any dependence of ΔA values from the incubation time, this parameter was raised to 6 min without any significant change.
The visible spectrum of LRO-loaded micelles prepared by method B (Fig. 2) suggests that (all-E)-lycopene is the sole micelle-associated carotenoid in this preparation letting its easy spectrophotometric quantitation as reported in Fig. 3. This allowed obtaining the TEAC value for pure (all-E)-lycopene incorporated in cholate/POPC micelles. For pure compounds TEAC was defined as the concentration of a Trolox solution with antioxidant potential equivalent to a 1 mM concentration of the molecule under investigation.44 This parameter is thus often given in mM units.45 However, being a relative parameter, we preferred the dimensionless expression (namely mmol of Trolox/mmol of antioxidant). (all-E)-Lycopene dispersed in POPC/cholate micelles exhibited an antioxidant power around 11 times higher than Trolox (TEAC = 10.8). A lower antioxidant capacity of (all-E)-lycopene (α-TEAC = 4) was recently reported by Muller et al.,46 who employed a two-phase water–n-hexane ABTS˙+ decolourization assay, analogue to that one adopted for LRO analysis. However these authors read the decolourization 2 min after lycopene/ABTS mixing, thus determining a “fast” α-TEAC value. This might partially explain the higher value obtained in our case, even though further factors are likely responsible for the enhanced activity of micelle-incorporated lycopene. Since the presence of oleoresin uncoloured components together with trace amount of different carotenoids is expected in micelles, a role of these co-incorporated compounds in amplifying the radical scavenging power of lycopene can be proposed.
The data collected by the ABTS˙+ decolourization assay were processed in order to evaluate the yield of antioxidant equivalents incorporation in small unilamellar vesicles. This investigation is particularly interesting since focused on the actual antioxidant capacity of final preparations regardless the chemical composition or composition changes occurring during the incorporation process. Degradation processes may affect negatively the antioxidant power, whilst the conversion to more reactive compounds or possible synergistic interactions may enhance the activity of incorporated compounds with respect to the original sample.
The results achieved are presented in Fig. 11 (upper histogram) and Table 1. The starting antioxidant amount (0.32 microequivalents of α-tocopherol) was contained in 20 mg of LRO. 25% of the initial antioxidant capacity was incorporated in micelles by method A, while the incorporation yield for method B was found slightly lower (21%). A further loss of α-tocopherol equivalents (α-TE) occurs upon the micelle to vesicle transition process, which appears particularly detrimental for micelles prepared by method B.
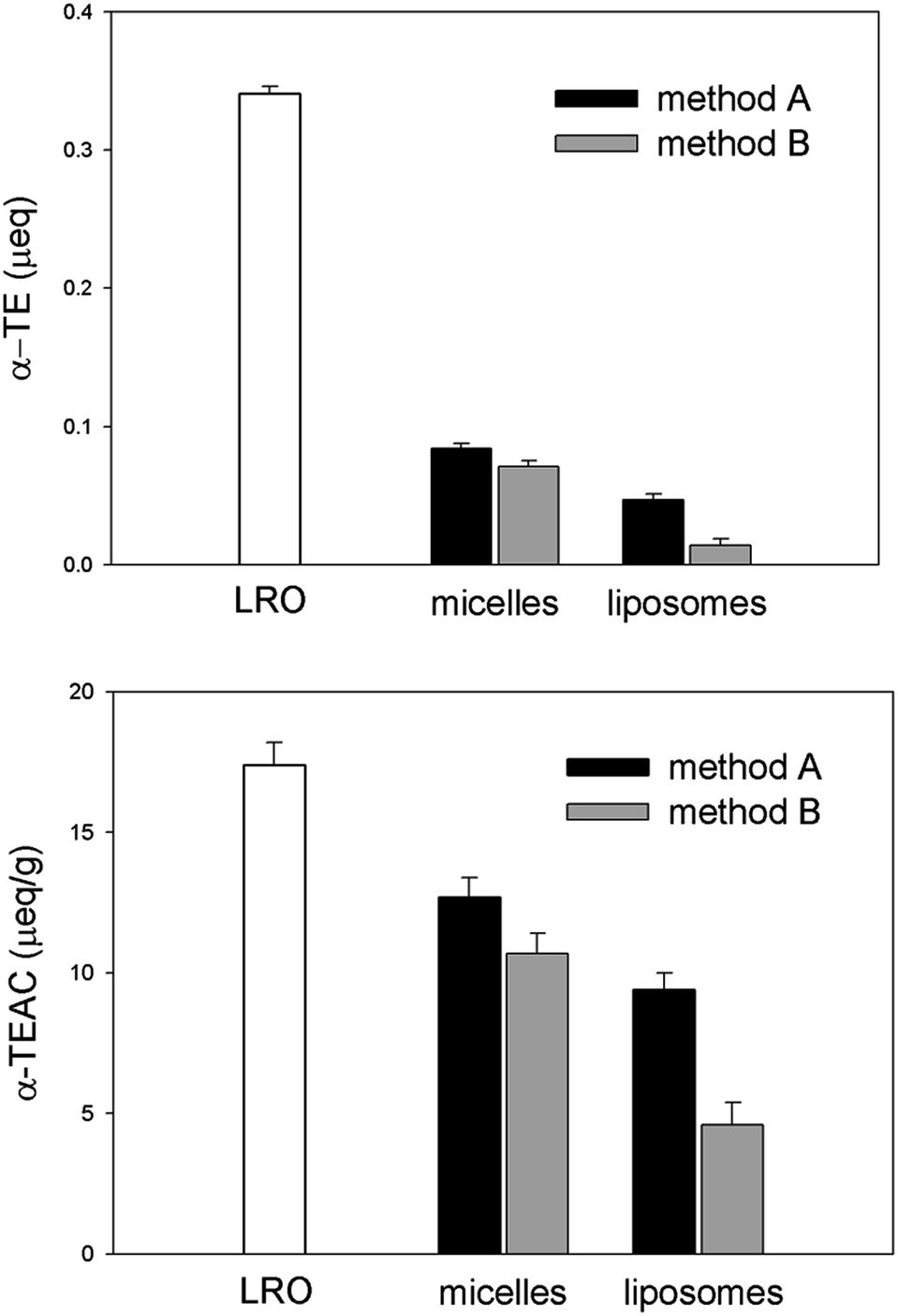 | ||
| Fig. 11 Upper panel: applied LRO α-TE amount (white histogram) and α-TE amount incorporated in mixed micelles and lipid vesicles for both methods A and B. Lower panel: specific antioxidant capacity of LRO and water-dispersed matrices relevant to both methods. Bulk POPC mass was used as reference for micelle and liposome samples. | ||
| Method A | Method B | |
|---|---|---|
| α-TE incorporation yield in micelles | 25% | 21% |
| α-TE incorporation yield in liposomes | 14% | 7% |
| Retained α-TEAC upon MVT | 56% | 32% |
The lower histogram in Fig. 11 depicts the antioxidant capacity of the different carotenoid-containing matrices expressed as α-TE per g. For micelle and liposome samples the bulk mass of lipids was taken as reference. Our data indicate that the specific antioxidant power of the water-dispersed liposome system, obtained by method A, is significantly high, representing 60% of that of the hydrophobic oleoresin. The goal of increasing the bio-availability of oleoresin carotenoids through water-soluble carriers was thus achieved with a limited reduction of specific radical-scavenging power. The transfer of antioxidant equivalents from an oil matrix to a vesicular carrying system resulted in a more significant decrease of specific antioxidant capacity when adopting method B for micelle preparation. This procedure, leading to the selective incorporation of (all-E)-lycopene appears however interesting in order to investigate the biological action of this stereoisomer.
Conclusions
The effective incorporation of hydrophobic antioxidants from LRO into small unilamellar liposomes by the MVT method was attained. The solvent-free characteristics of the original oleoresin were preserved by avoiding the use of chloroform for liposome preparation, which represents a fundamental ingredient in the original protocol.22 Chloroform is indeed an excellent solvent for both lipids and almost all hydrophobic compounds of biological interest. The employment of chloroform allows to easily and precisely control the ratio of the vesicle components and its subsequent evaporation leads to the effective dispersion of hydrophobic molecules into the lipid matrix. The homogeneity of the dry layer interacting with the detergent plays a key role in achieving the proper dispersion of desired molecules into mixed micelles. The substitution of chloroform with ethanol (method A) resulted in a low incorporation yield of (all-E)-lycopene, in agreement with its poor solubility in ethanol. Visible spectra and antioxidant activity assessment revealed however a significant content of various carotenoids, which exhibit interesting radical scavenging properties. On the other hand pre-formed cholate/POPC micelles, applied to the oleoresin (method B), proved to extract selectively (all-E)-lycopene, incorporating only negligible amount of different stereoisomers and carotenoids. In this case the overall loaded amount of bio-active compounds was lower as well as the specific antioxidant activity. Nevertheless method B appears extremely interesting aiming to investigate (all-E)-lycopene activity and to scale up the preparation. The procedure is in fact easy, rapid, and suitable for treating large quantity of oleoresins. Ethanol is not employed making the procedure absolutely solvent-free and cheap. Moreover the possibility of running multiple extractions on the same oleoresin sample would avoid wasting of material improving the process efficiency.Both methods A and B produced homogeneous populations of liposomes with a 40 nm diameter, containing negligible amount of oleoresin glycerides, indicating that mixed micelles act as extracting agents of bioactive compounds.
The analysis of ABTS˙+ attenuation and decolourization curves revealed that antioxidant potential of vesicle-dispersed carotenoids do not act as a Trolox-like mechanism. In particular the “total” TEAC was found higher than the “fast” TEAC indicating that the ABTS˙+ neutralization reaction produces carotenoid-derivatives which are in turn radical scavengers, although with a kinetics slower than that one of parent compounds.
Our ABTS˙+ decolourization measurements with trolox and micelle-incorporated α-tocopherol demonstrated that comparison between data obtained in organic and aqueous solvents is feasible, thus allowing the reliable evaluation of antioxidant equivalents incorporation yields. It is indeed well known that different reaction media often generate conflicting results in the determination of antioxidant power of bio-active compounds. Nevertheless it will be interesting to investigate the capacity of incorporated carotenoids to scavenge peroxyl radicals, rather than non-physiological radicals such as ABTS˙+. In this regard the reliability for carotenoids analysis of an oxygen radical absorbance capacity (ORAC) assay employing a fluorescent probe and acetonitrile, 2,2′-azobis(isobutyronitrile) (AIBN) as radical generator has been recently reported.47
Nutraceutical and pharmaceutical potential of these SUV preparations may be improved testing other lipids and lipid mixtures. The structure of lipids might indeed affect significantly the loading capability of micelles and vesicles and plays a significant role in establishing interactions with cell membranes. As an example the surface charge of liposomes was demonstrated to strongly affect their binding and endocytosis in different cell lines.48 Moreover the decoration of liposomal surface by attaching specific ligands49 can be achieved easily by the MVT method in order to increase intracellular drug levels in target areas. Hence further investigations will be addressed to improve our formulations for optimizing the liposome-mediated delivery of LRO carotenoids to intracellular targets and to elucidate their actual protective action against biological radicals.
On the other hand, any natural hydrophobic extract can be virtually incorporated in liposomes by means of the mixed micelles tool, in order to make bio-deliverable its bio-activity. The general application of our approach is very interesting since drug-loaded liposomes are usually produced using pure bioactive compounds, which are often expensive and/or unstable. This work demonstrates instead that the versatile MVT method can be adapted to mixtures of oil-solubilized hydrophobic compounds, avoiding chloroform and any other toxic organic solvent, thus reducing preparation costs and increasing biocompatibility and bio-activity of the final formulations.
Acknowledgements
This work has been financially supported by the Italian research funding programs: PON 254/Ric., cod. PONa3_00334 (2HE Center for Human Health and Environment), PON Pro.Ali.Fun cod. PON02_00186_2937475, Industria 2015 (Nuove Tecnologie per il Made in Italy “Produzione ed applicazioni di licopene biologico”), and by Regione Puglia (“Ritorno al Futuro” and “Sens&Micro LAB Project (2007–2013)” grants). The authors thank Francesca Vedruccio, Paola Antonazzo and Davide Pantaleo for their contribution to the experimental work.Notes and references
- R. Amarowicz, Eur. J. Lipid Sci. Technol., 2011, 113, 675–677 CrossRef CAS
.
- P. Di Mascio, S. Kaiser and H. Sies, Arch. Biochem. Biophys., 1989, 274, 532–538 CrossRef CAS
- F. Khachik, L. Carvalho, P. S. Bernstein, G. J. Muir, D. Y. Zhao and N. B. Katz, Exp. Biol. Med., 2002, 227, 845–851 CAS
- L. L. Tang, T. Y. Jin, X. B. Zeng and J. S. Wang, J. Nutr., 2005, 135, 287–290 CAS
- E. Gouranton, C. Thabuis, C. Riollet, C. Malezet-Desmoulins, C. El Yazidi, M. J. Amiot, P. Borel and J. F. Landrier, J. Nutr. Biochem., 2011, 22, 642–648 CrossRef CAS PubMed
- Y. P. Zhao, W. L. Yu, W. L. Hu and Y. Ying, Nutr. Res., 2003, 23, 1591–1595 CrossRef CAS
- G. Riccioni, B. Mancini, E. Di Ilio, T. Bucciarelli and N. D'Orazio, Eur. Rev. Med. Pharmacol. Sci., 2008, 12, 183–190 CAS
- A. W. Williams, T. W. M. Boileau and J. W. Erdman, Proc. Soc. Exp. Biol. Med., 1998, 218, 106–108 CrossRef CAS
- D. V. Ratnam, D. D. Ankola, V. Bhardwaj, D. K. Sahana and M. N. V. R. Kumar, J. Controlled Release, 2006, 113, 189–207 CrossRef CAS PubMed
- R. Salvayre, N. Lauth-de Viguerie and J.-D. Marty, Trends Biotechnol., 2012, 30, 485–496 CrossRef PubMed
- C. Banville, J. C. Vuillemard and C. Lacroix, Int. Dairy J., 2000, 10, 375–382 CrossRef CAS
- A. K. Thompson, A. Couchoud and H. Singh, Dairy Sci. Technol., 2009, 89, 99–113 CrossRef CAS PubMed
- C. Tan, J. Xue, X. Lou, S. Abbas, Y. Guan, B. Feng, X. Zhang and S. Xia, Food Funct., 2014, 5, 1232–1240 CAS
- A. Hentschel, S. Gramdorf, R. H. Müller and T. Kurz, J. Food Sci., 2008, 73, N1–N6 CrossRef CAS PubMed
- I. Lancrajan, H. A. Diehl, C. Socaciu, M. Engelke and M. Zorn-Kruppa, Chem. Phys. Lipids, 2001, 112, 1–10 CrossRef CAS
- C. Socaciu, P. Bojarski, L. Aberle and H. A. Diehl, Biophys. Chem., 2002, 99, 1–15 CrossRef CAS
- C. Socaciu, R. Jessel and H. A. Diehl, Chem. Phys. Lipids, 2000, 106, 79–88 CrossRef CAS
- A. A. Woodall, G. Britton and M. J. Jackson, Biochim. Biophys. Acta, 1997, 1336, 575–586 CrossRef CAS
- H. P. McNulty, J. Byun, S. F. Lockwood, R. F. Jacob and R. P. Mason, Biochim. Biophys. Acta, 2007, 1768, 167–174 CrossRef CAS PubMed
- J. Liang, Y. X. Tian, F. Yang, J. P. Zhang and L. H. Skibsted, Food Chem., 2009, 115, 1437–1442 CrossRef CAS PubMed
- M. S. Lenucci, A. Caccioppola, M. Durante, L. Serrone, L. Rescio, G. Piro and G. Dalessandro, J. Sci. Food Agric., 2010, 90, 1709–1718 CrossRef CAS PubMed
- M. Ollivon, S. Lesieur, C. Grabielle-Madelmont and M. Paternostre, Biochim. Biophys. Acta, 2000, 1508, 34–50 CrossRef CAS
- M. Holzer, S. Barnert, J. Momm and R. Schubert, J. Chromatogr. A, 2009, 1216, 5838–5848 CrossRef CAS PubMed
- D. Liu, A. Mori and L. Huang, Biochim. Biophys. Acta, 1992, 1104, 95–101 CrossRef CAS
- P. L. Beaumier and K. J. Hwang, Biochim. Biophys. Acta, 1983, 731, 23–30 CrossRef CAS
- G. Song, H. L. Wu, K. Yoshino and W. C. Zamboni, J. Liposome Res., 2012, 22, 177–192 CrossRef CAS PubMed
- R. Re, N. Pellegrini, A. Proteggente, A. Pannala, M. Yang and C. Rice-Evans, Free Radical Biol. Med., 1999, 26, 1231–1237 CrossRef CAS
- C. Rice-Evans and N. J. Miller, in Methods Enzymol, ed. P. Lester, Academic Press, 1994, vol. 234, pp. 279–293 Search PubMed
- T. W. Yu and C. N. Ong, Anal. Biochem., 1999, 275, 217–223 CrossRef CAS PubMed
- N. J. Miller, J. Sampson, L. P. Candeias, P. M. Bramley and C. A. Rice-Evans, FEBS Lett., 1996, 384, 240–242 CrossRef CAS
- A. Agostiano, F. Milano and M. Trotta, Photosynth. Res., 2005, 83, 53–61 CrossRef CAS PubMed
- F. Milano, F. Italiano, A. Agostiano and M. Trotta, Photosynth. Res., 2009, 100, 107–112 CrossRef CAS PubMed
- S. Twomey, J. Comput. Phys., 1975, 18, 188–200 CrossRef
- L. Zechmeister, A. L. LeRosen, W. A. Schroeder, A. Polgár and L. Pauling, J. Am. Chem. Soc., 1943, 65, 1940–1951 CrossRef CAS
- M. Takehara, M. Nishimura, T. Kuwa, Y. Inoue, C. Kitamura, T. Kumagai and M. Honda, J. Agric. Food Chem., 2013, 62, 264–269 CrossRef PubMed
- C. Longo, L. Leo and A. Leone, Int. J. Mol. Sci., 2012, 13, 4233–4254 CrossRef CAS PubMed
- V. De Leo, L. Catucci, A. Falqui, R. Marotta, M. Striccoli, A. Agostiano, R. Comparelli and F. Milano, Langmuir, 2014, 30, 1599–1608 CrossRef CAS PubMed
- C. P. Baron and H. J. Andersen, J. Agric. Food Chem., 2002, 50, 3887–3897 CrossRef CAS PubMed
- M. J. T. J. Arts, J. Sebastiaan Dallinga, H.-P. Voss, G. R. M. M. Haenen and A. Bast, Food Chem., 2003, 80, 409–414 CrossRef CAS
- M. J. T. J. Arts, J. Sebastiaan Dallinga, H.-P. Voss, G. R. M. M. Haenen and A. Bast, Food Chem., 2004, 88, 567–570 CrossRef CAS PubMed
- M. J. T. J. Arts, G. R. M. M. Haenen, H.-P. Voss and A. Bast, Food Chem. Toxicol., 2004, 42, 45–49 CrossRef CAS PubMed
- R. van den Berg, G. R. M. M. Haenen, H. van den Berg, W. van der Vijgh and A. Bast, Food Chem., 2000, 70, 391–395 CrossRef CAS
- D. Villano, M. S. Fernandez-Pachon, A. M. Troncoso and M. C. Garcia-Parrilla, Talanta, 2004, 64, 501–509 CrossRef CAS PubMed
- C. A. Rice-Evans, N. J. Miller and G. Paganga, Free Radicals Biol. Med., 1996, 20, 933–956 CrossRef CAS
- V. Bohm, N. L. Puspitasari-Nienaber, M. G. Ferruzzi and S. J. Schwartz, J. Agric. Food Chem., 2002, 50, 221–226 CrossRef PubMed
- L. Muller, P. Goupy, K. Frohlich, O. Dangles, C. Caris-Veyrat and V. Bohm, J. Agric. Food Chem., 2011, 59, 4504–4511 CrossRef PubMed
- E. Rodrigues, L. R. B. Mariutti, R. C. Chiste and A. Z. Mercadante, Food Chem., 2012, 135, 2103–2111 CrossRef CAS PubMed
- C. R. Miller, B. Bondurant, S. D. McLean, K. A. McGovern and D. F. O'Brien, Biochemistry, 1998, 37, 12875–12883 CrossRef CAS PubMed
- R. R. Sawant and V. P. Torchilin, Soft Matter, 2010, 6, 4026–4044 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Normalized absorption spectra relevant to pigments extracted from LRO-loaded mixed micelles prepared by method A, and time evolution of ABTS˙+ decolourization obtained for standard α-tocopherol and LRO. See DOI: 10.1039/c4ra12254b |
| This journal is © The Royal Society of Chemistry 2015 |