
Aliphatic thermoplastic polyurethane-ureas and polyureas synthesized through a non-isocyanate route†
Abstract
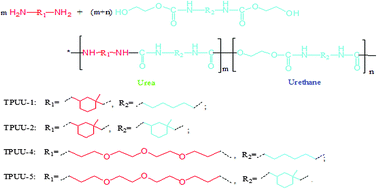
A simple non-isocyanate route for synthesizing aliphatic thermoplastic polyurethane-ureas (TPUUs) and thermoplastic polyureas (TPUreas) is presented. Melt transurethane polycondensation of isophorone diamine or diethylene glycol bis(3-aminopropyl) ether with bis(hydroxyethyl) hexanediurethane, bis(hydroxyethyl) isophoronediurethane or bis(hydroxyethyl) piperazinediurethane was conducted at 170 °C under a reduced pressure of 3 mmHg. A series of thermoplastic TPUUs and TPUreas were prepared, and were characterized by gel permeation chromatography, FT-IR, 1H-NMR, differential scanning calorimetry, thermogravimetric analysis, and tensile testing. The TPUUs and TPUreas have an Mn up to 14 900 g mol−1, an Mw up to 43 700 g mol−1, Tg between −18.6 °C and 116.8 °C, and an initial decomposition temperature of over 222.3 °C. A flexible TPUU exhibits a melting temperature of 77.7 °C, a tensile strength of 6.46 MPa, and an elongation at break of 180.20%.
 Please wait while we load your content...
Please wait while we load your content...

