
Preparation and characterization of activated montmorillonite clay supported 11-molybdo-vanado-phosphoric acid for cyclohexene oxidation
Abstract
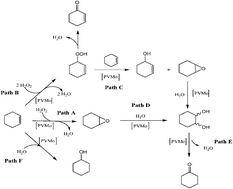
A new heterogeneous catalyst (PVMo/bentonite), consisting of vanadium substituted polyphosphomolybdate with Keggin structure H4[PVMo11O40]·13H2O (PVMo) supported on acid activated bentonite (clay from Hammam Boughrara, Maghnia, Algeria), was prepared by witness impregnation and characterized by X-ray diffraction, BET, Fourier-transformed infrared spectroscopy, 31P NMR, UV-vis diffuse reflectance spectroscopy and thermogravimetric & differential thermal analysis (TG-DTA). X-ray diffraction indicated that PVMo was properly loaded on bentonite as a support. Heterogenization of homogenous catalysts is really interesting, as heterogeneous catalysts are recoverable. Therefore, the synthesized materials can be used as efficient heterogeneous catalysts for epoxidation of cyclohexene. The obtained results showed that a better catalytic activity can be obtained with PVMo/bentonite (81.5% of conversion), by drop addition of H2O2, for 3 hours.
 Please wait while we load your content...
Please wait while we load your content...

