
A practical, catalytic and selective deprotection of a Boc group in N,N′-diprotected amines using iron(iii)-catalysis†
Abstract
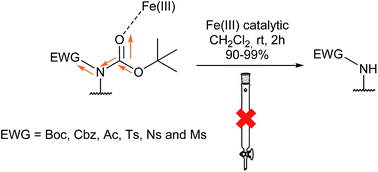
A selective, catalytic and practical method for removing a Boc group from several N,N′-diprotected amino acids and amine derivatives using iron(III) salts as sustainable catalysts is described. The process is clean, not needing a purification step. A theoretical study rationalizing the results with several metals is presented.
 Please wait while we load your content...
Please wait while we load your content...

