
Design of efficient sergeant molecules for chiral induction in nano-porous supramolecular assemblies†
Andrea Minoia*a,
Iris Destoopb,
Elke Ghijsensb,
Steven De Feyterb,
Kazukuni Taharac,
Yoshito Tobec and
Roberto Lazzaronia
aUniversity of Mons – UMONS, Place du Parc 20, 7000 Mons, Belgium. E-mail: andrea.minoia@umons.ac.be; Tel: +32 6537 3859
bDepartment of Chemistry, Division of Molecular and Nano Materials, KUL, Celestijnenlaan 200F, 3001 Leuven, Belgium
cDivision of Frontier Materials Science, Graduate School of Engineering Science, Osaka University, Toyonaka, Osaka 560-8531, Japan
First published on 17th December 2014
Abstract
In this study molecular modeling is used to investigate the transfer of chirality from the molecular level to homochiral, nano-porous 2D networks in self-assembled monolayers at the liquid/graphite interface. This allows proposing design rules for sergeant molecules for inducing chiral supramolecular assembly in nano-porous monolayers.
The induction and the control of chirality in 2-dimensional self-assembled monolayers on surfaces is a topic of great interest, both from the experimental and the modeling point of view.1 In particular, the possibility to form 2-dimensional nano-porous supramolecular assemblies of specific chirality is essential for the adsorption of guest molecules into the 2D network based on size and shape selectivity.2 Alkoxylated dehydrobenzo[12]annulene (DBA) derivatives (Fig. 1a) are known to self-assemble at the liquid/HOPG graphite interface and form nanoporous networks in which the molecules arrange in chiral honeycomb porous structures (Fig. 1b). In those layers, the alkoxy side groups can interdigitate in two different ways: −Type or +Type (Fig. 1c).3 This in turn induces the formation of pores with a well-defined handedness: −Type interdigitation leads to the formation of clockwise (CW) pores, while +Type interdigitation leads to the formation of counterclockwise (CCW) pores (Fig. 1d). When the DBA molecule is achiral and the monolayer is formed from an achiral solvent, such as 1-octanol, a racemic monolayer (the CW
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CCW ratio is 1:1) is observed in the STM images. Interestingly, CW and CCW pores do not mix, but rather form homochiral domains (see a Scanning Tunneling Microscopy – STM-image in Fig. 1b). When the DBA molecule contains stereogenic centers, preferential formation of one type of pore is observed.4 In this work, we use molecular modeling to systematically investigate the relation between the molecular structure of the building blocks and the resulting chiral induction in the nano-porous monolayer when different DBA molecules co-assemble at the surface. The aim of this work is thus to understand, through molecular modeling simulations, how chirality at the molecular level is transferred at the selection of sergeant molecules to induce a specific chirality in supramolecular self-assemblies, by efficiently exploiting the well-known sergeant–soldier mechanism. In this work we focus on the promotion of chirality in nano-porous DBA assemblies induced by five different DBA sergeant molecules. In practice, we consider as a model system the elementary structural unit of those 2D networks, i.e., a single CW or CCW pore adsorbed on a HOPG graphite substrate, which is kept frozen. The chiral bias is induced by exchanging one achiral DBA molecule of the network with a different DBA molecule that will act as a candidate sergeant molecule. The efficiency of the sergeant molecule in inducing a specific chirality in the supramolecular assembly is related to its capacity to stabilize one chiral honeycomb structure, say CW, over the CCW one. This hypothesis was supported by molecular mechanics calculations, using structure optimization.2 The reason for considering a single pore as the structural element for the modeling lies in the fact that it is known experimentally that CW and CCW pores do not mix, but, instead, do form separate homochiral domains: once a pore of a particular chirality is formed, it is energetically favorable for the molecules next to it to aggregate according to its chirality rather than aggregating against it.
:CCW ratio is 1:1) is observed in the STM images. Interestingly, CW and CCW pores do not mix, but rather form homochiral domains (see a Scanning Tunneling Microscopy – STM-image in Fig. 1b). When the DBA molecule contains stereogenic centers, preferential formation of one type of pore is observed.4 In this work, we use molecular modeling to systematically investigate the relation between the molecular structure of the building blocks and the resulting chiral induction in the nano-porous monolayer when different DBA molecules co-assemble at the surface. The aim of this work is thus to understand, through molecular modeling simulations, how chirality at the molecular level is transferred at the selection of sergeant molecules to induce a specific chirality in supramolecular self-assemblies, by efficiently exploiting the well-known sergeant–soldier mechanism. In this work we focus on the promotion of chirality in nano-porous DBA assemblies induced by five different DBA sergeant molecules. In practice, we consider as a model system the elementary structural unit of those 2D networks, i.e., a single CW or CCW pore adsorbed on a HOPG graphite substrate, which is kept frozen. The chiral bias is induced by exchanging one achiral DBA molecule of the network with a different DBA molecule that will act as a candidate sergeant molecule. The efficiency of the sergeant molecule in inducing a specific chirality in the supramolecular assembly is related to its capacity to stabilize one chiral honeycomb structure, say CW, over the CCW one. This hypothesis was supported by molecular mechanics calculations, using structure optimization.2 The reason for considering a single pore as the structural element for the modeling lies in the fact that it is known experimentally that CW and CCW pores do not mix, but, instead, do form separate homochiral domains: once a pore of a particular chirality is formed, it is energetically favorable for the molecules next to it to aggregate according to its chirality rather than aggregating against it.
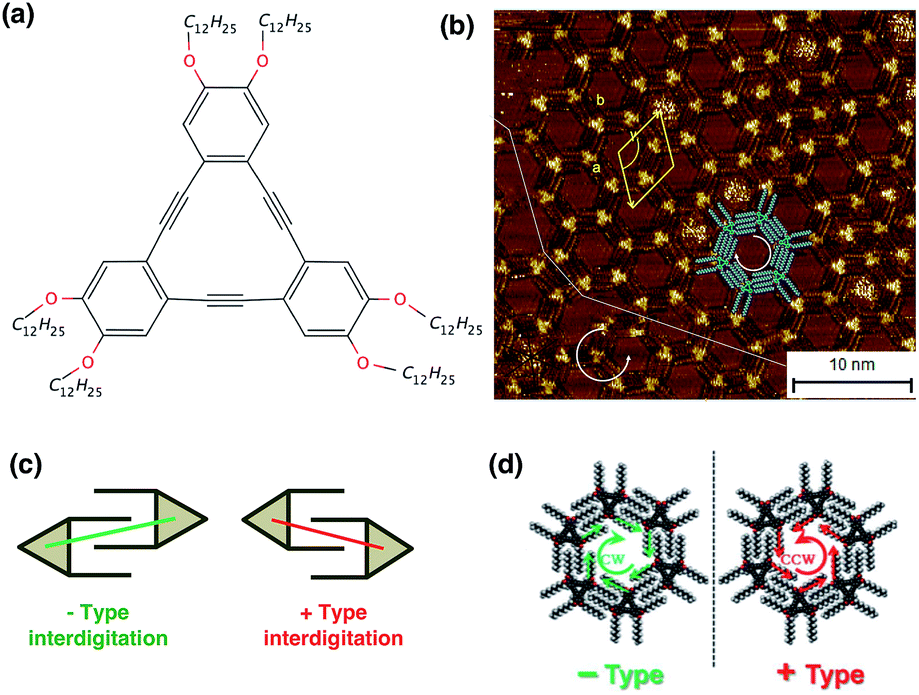 | ||
| Fig. 1 (a) Chemical structure for the alkoxylated dehydrobenzo[12]annulene DBA-OC12 molecule; (b) STM image of a DBA-OC12 monolayer; (c) scheme of the two possible (−Type and +Type) interdigitations between DBA molecules; (d) CW and CCW pores formed by 6 interdigitated DBA molecules. | ||
Despite the good match between experiment and the early molecular mechanics approach,1 important questions remain open on the molecular origin of the chiral induction and on the relative stability of the two different types of pores. From the methodological point of view, one of the main challenges is to select an efficient procedure to determine the most stable structures for the enantiomeric CW and CCW pores. For this reason, we have designed and implemented a more advanced and specific MD/Quench approach, which includes a structure-recombination (SR) step to generate new, independent, structures that are sampled in the attempt to locate the lowest energy structure of the pores. Next, a detailed energetic and structural analysis is required in order to pinpoint the relation between the molecular structure of the sergeant molecule and the chiral induction in the assembly.
In a previous work5 aimed at the understanding of the chiral expression in windmill-like self-assembled structures of phenylene vinylene oligomers at the liquid/HOPG interface, we have introduced the iterative Molecular Dynamics (MD)/Quench procedure to obtain the most stable CW and CCW structures. This approach works well for self-assemblies in which the molecules strongly interact between them (e.g., via hydrogen bonding), so that the molecular aggregates remain stable under the use of the high-temperature MD simulations needed to properly explore large regions of the potential energy landscape of the system. However, the DBA structures considered here are formed by interdigitating DBAs alkoxy groups and the pore stability is based only on the relatively weak van der Waals interactions between the interdigitated chains. It was therefore essential to improve the modeling approach, via the MD/Quench + SR scheme, to obtain a meaningful energetic and topological comparison between the two chiral structures. The new approach works as following: the initial guess geometry of the modelled assembly is built and optimized on a frozen HOPG surface. Next, a 150 ps-long MD is performed in the NVT ensemble at 500 K in order to sample different orientations of the assembly on the surface. The initial high-energy MD simulation generates 300 structures that are all fully minimized and their energies are analyzed and compared. From this point on, the search for the most stable structure is done iteratively. The Structure Recombination (SR) step consists in aligning the lowest-energy structure for the pore alone to the pore geometry that best interacts with the surface. This SR structure, which is uncorrelated to those obtained from the previous MD simulation, is then minimized. This helps to better explore the potential energy landscape of the system. To further refine the search of the lowest-energy structure, both the lowest total potential energy structure found during the previous MD simulation and the SR structure are annealed via a 30 ps-long MD simulation, during which the temperature is lowered to 0 K in a sigmoidal way. Each annealing simulation produces 60 structures, which are all minimized and their energy analyzed and compared. A new low-energy structure is found and a new SR structure is created and quenched. These new structures are annealed and the whole procedure is repeated until the convergence criterium for the energy is reached, i.e. when the improvement of the energy of the system is smaller than 0.01 kcal mol−1. The lowest-energy structures obtained for the CW and CCW pores are considered to be the most stable aggregates on the surface and are used to investigate the role of the sergeant molecule on their stability. It is important to note here that the CW and CCW distinction is meaningful when considering the orientation of the molecules with respect to the surface underneath: a CW pore, when flipped over on the surface, will become CCW. Therefore, in principle there are two distinct ways to turn a CW pore into a CCW pore: the first one consists in changing the way all the molecules interdigitate, while the second one involves flipping a CW pore upside/down on the surface to obtain a CCW pore, named Flipped_CW (FCW). In the same way, flipping a CCW pore upside/down on the surface will result in a CW pore, named Flipped_CCW (FCCW). The four possible structures for a DBA-OC12 pore, i.e., CW, CCW, FCW and FCCW pores, are shown in Fig. 2. In order to highlight the different interdigitation, we coloured in red the alkoxy chains attached to the left side of a DBA core and in blue those attached to the right side. The black arrows in the middle of the pores show the handedness (or sense of rotation) of the pores. As example, both CW and FCCW pores have the same clockwise handedness, but their molecules have different interdigitation: in the CW pore, the red alkoxy groups define the border of the outer perimeter of the pore, while in the FCCW that perimeters is defined by blue alkoxy groups. Depending on the symmetry of the molecules involved, the CW (CCW) and FCCW (FCW) pores can be equivalent.
 | ||
| Fig. 2 CW, CCW, FCW and FCCW DBA-OC12 pores. Alkoxy groups on the left side of the DBA cores are coloured in red, while those attached to the right side of the DBA cores are in blue. | ||
For this study, we used the TINKER6 molecular modeling package and its MM3 force field,7 which properly describes the type of interactions acting in these systems. As a first step, we validated the modeling protocol on CW and CCW pores formed by six achiral DBA-OC12 (Fig. 1a) molecules, which are known to exist in a 50:50 ratio in the absence of any chiral bias. We find that the most stable structures for the CW and CCW pores of achiral DBA-OC12 molecules are isoenergetic, having both a total potential energy on the surface of −353.2 kcal mol−1, consistent with the 50:50 ratio observed experimentally. This agreement between modeling and experimental results supports the validity of the methodology we have developed. We also compute the binding energy, BE, of the system, which estimates the energy gained (or lost) by the molecules when incorporated in the supramolecular self-assembly on the surface, with respect to the total energy of the molecules when they are not interacting with one another, i.e., when each molecule is isolated. The BE is obtained using eqn (1).
| BE = Esys − (5Esoldier + Esergeant) | (1) |
 | ||
| Fig. 3 Chemical structures of the DBA derivatives theoretically investigated in this work. | ||
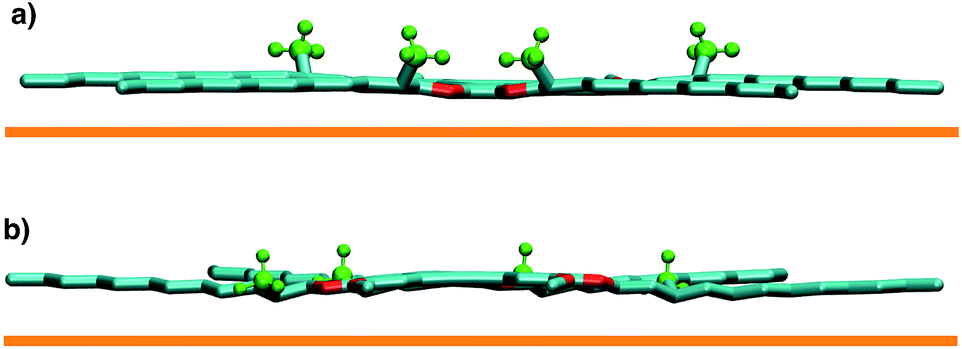 | ||
| Fig. 4 Molecular conformations for a single DBA-OC12(2-S)-OC12(2-R) molecule adsorbed on graphite (represented by the orange line) with the methyl groups attached to the stereogenic centres (in green) pointing (a) away and (b) towards the surface. Hydrogen atoms other than those on the methyl groups are not shown for the sake of clarity. | ||
The modified MD/Quench simulations indicate that the CW and CCW pores are isoenergetic, having both a total potential energy on the surface of −348.3 kcal mol−1 (see Table 1 in ESI†). This is consistent with the fact that there is no chirality at the molecular level that can be transferred to the assembly, thus favoring one pore's chirality over the other. Because our goal is to design molecules that can induce a chirality in the monolayer, this first candidate is not appropriate. We thus modified the molecular structure by displacing every other stereogenic center by one CH2 unit along the alkoxy chain, so as to generate only (S)- centers in the molecule, which is now chiral. We will refer to this new molecule as cDBA-OC12(2-S)-OC12(3-S). As in the previous compound, the molecular up/down symmetry is broken due to the presence of the methyl groups attached to the stereogenic centers (as above, we will consider only CW and CCW pores, for which those methyl groups are pointing away from the surface). However the equivalence between +Type and −Type interdigitation is also lost, because of the presence of two different alkoxy groups in the molecule. The results from the energetic analysis show that the CW pore is about 0.7 kcal mol−1 more stable than its CCW counterpart (see Table 2 in ESI†). This indicates that the sergeant molecule could promote a specific chirality for the pore. In order to check if such sergeant molecule can assemble with the DBA-OC12 soldier molecules, we compute the molecular binding energies for the CW and CCW structures (with five soldier molecules and one sergeant molecule). The resulting BE are −95.4 kcal mol−1 and −94 kcal mol−1 for the CW and CCW pore, respectively, which are comparable to those obtained for the DBA-OC12 pores (−94.5 kcal mol−1). This indicates that sergeant and soldier molecules can indeed self-assemble to form such “mixed” pore. Because the extra stability of the CW pore over the CCW one is 0.7 kcal mol−1, comparable to the thermal energy, kBT, at room temperature (0.6 kcal mol−1), the cDBA-OC12(2-S)-OC12(3-S) sergeant molecule is not expected to be strongly efficient in inducing the formation of a homochiral, CW, monolayer.
Another way to try to induce chirality is to consider a sergeant molecule having alternating alkoxy groups of different length, e.g., –OC12H25 and –OC13H27 (DBA-OC12-OC13). Because such molecule does not have stereogenic centers, the up/down symmetry is maintained, while +Type and −Type interdigitations are not equivalent due to the different length of the alkoxy groups in the sergeant molecule: in the −Type interdigitation (CW pore), the –OC13H27 alkoxy groups lie along the inner and outer perimeter of the pore, while in the +Type interdigitation (CCW pore), those groups are sandwiched between the two –OC12H25 alkoxy groups from the adjacent DBA-OC12 molecules (Fig. 5).
 | ||
| Fig. 5 (a) CW and (b) CCW DBA-assemblies with one DBA-OC12-OC13 sergeant molecule. The –OC13H27 alkoxy groups of the sergeant molecule are shown in green. | ||
The result is that four pores can be built, which reduce to two different situations because CW is equivalent to FCW, and CCW is equivalent to FCCW. We find that the pores based on −Type interdigitations are 4.4 kcal mol−1 more stable than those based on +Type interdigitations (see Table 3 in ESI†). This difference in stability is mostly due to the stress introduced in the structure of the +Type-based pore by having the longer alkoxy groups fully interdigitated with those of the neighbor DBA-OC12 molecules, which results in a significant deformation of the CCW honeycomb geometry. The stabilization achieved for the −Type-based pore over the +Type counterpart is large, around 8kBT, but the DBA-OC12-OC13 molecule is not expected to promote the formation of a chiral monolayer because of the equivalence between CW and FCW, due to the up/down symmetry of the sergeant molecule.
The DBA-OC12-OC13 molecule appears to be the ideal sergeant molecule if its up/down symmetry can be broken, so that CW and FCW pores are no longer equivalent. Inspired by the DBA-OC12(2-S)-OC12(2-R) and cDBA-OC12(2-S)-OC12(3-S) molecules, the simplest way to break the up/down symmetry is to introduce stereogenic centers in the molecule. Introducing stereogenic centers in the cDBA-OC12-OC13 molecule leads to the cDBA-OC12(2-S)-OC13(2-R) molecule. A single cDBA-OC12(2-S)-OC13(2-R) molecule adsorbed on the HOPG surface with the methyl groups attached to the stereogenic centers pointing upwards is 11.3 kcal mol−1 more stable than when it is adsorbed with those groups pointing to the surface. This means that not only −Type and +Type-based pores are not equivalent (because of the different length of the alkoxy groups), but now also that CW and CCW pores based on the same interdigitation scheme are not equivalent, because of the disruption of the up/down symmetry. The result is that the CW pore is 2.9 kcal mol−1 more stable than the CCW counterpart (see Table 4 in ESI†). This stabilisation is of the order of 5kBT, which indicates that the molecule is indeed able to induce the selective formation of CW pores. The difference in stability is lower than that promoted by the DBA-OC12-OC13 molecule (4.4 kcal mol−1), probably due to the stress introduced in the pore by the presence of the methyl groups attached to the stereogenic centers in the cDBA-OC12(2-S)-OC13(2-R) molecule. Nevertheless, the cDBA-OC12(2-S)-OC13(2-R) molecule is an efficient sergeant molecule to promote the formation of CW chiral nanoporous monolayers, which allows rationalising what is observed experimentally.4
In conclusion, we have presented a systematic modeling approach to the design of efficient sergeant molecules for the formation of self-assembled monolayers of the desired chirality. Because the results are based on the relative stability of the chiral forms of assemblies of the building blocks (pores), we developed a new modeling strategy to efficiently locate the most stable structure for self-assembled monolayers in which molecules interact only via weak van der Waals and electrostatic interactions. Here the efficiency of the chiral induction promoted in the structural unit of the 2D-nano-porous DBA assembly by the candidate sergeant molecule has been investigated by systematically tuning the molecular structure of the sergeant, so that the most efficient sergeant compound has been identified. This methodology could be extended to the general design of sergeant molecules for chiral supramolecular assembly for which a well-defined 2D structural unit (or unit cell) can be identified.
Acknowledgements
The Mons-Leuven collaboration is supported by the Science Policy Office of the Belgian Federal Government (IAP 7/05). Research in Mons is also supported by FRS-FNRS and Région Wallonne (OPTI2MAT excellence program). Research in Leuven is supported by the Fund of Scientific Research Flanders (FWO), KU Leuven (GOA 11/003), and the European Research Council under the European Union's Seventh Framework Programme (FP7/2007-2013)/ERC Grant Agreement no. 340324.Notes and references
- S. De Feyter and F. C. De Schryver, Chem. Soc. Rev., 2003, 32, 139 RSC; M. Linares, A. Minoia, P. Brocorens, D. Beljonne and R. Lazzaroni, Chem. Soc. Rev., 2009, 38, 806 RSC; A. Minoia, Z. Guo, H. Xu, S. J. George, A. P. H. J. Schenning, S. De Feyter and R. Lazzaroni, Chem. Commun., 2011, 47, 10924 RSC.
- G. Schull, L. Douillard, C. Fiorini-Debuisschert and F. Charra, Nano Lett., 2006, 6, 1360 CrossRef CAS PubMed; J. Adisoejoso, et al., Angew. Chem., Int. Ed., 2009, 48, 7353 CrossRef PubMed.
- K. Tahara, S. Furukawa, H. Uji-I, T. Uchino, T. Ichikawa, J. Zhang, W. Mamdouh, M. Sonoda, F. C. De Schryver, S. De Feyter and Y. Tobe, J. Am. Chem. Soc., 2006, 128, 16613 CrossRef CAS PubMed; S. Lei, K. Tahara, F. C. De Schryver, M. Van der Auweraer, Y. Tobe and S. De Feyter, Angew. Chem., Int. Ed., 2008, 47, 2964 CrossRef PubMed; K. Tahara, S. Lei, J. Adisoejoso, S. De Feyter and Y. Tobe, Chem. Commun., 2010, 46, 8507 RSC.
- K. Tahara, H. Yamaga, E. Ghijsens, K. Inukai, J. Adisoejoso, M. O. Blunt, S. De Feyter and Y. Tobe, Nat. Chem., 2011, 3, 714 CrossRef CAS PubMed.
- A. Minoia, Z. Guo, H. Xu, S. J. George, A. P. H. J. Schenning, S. De Feyter and R. Lazzaroni, Chem. Commun., 2011, 47, 10924 RSC.
- http://dasher.wustl.edu/tinker/.
- J.-H. Lii and N. L. Allinger, J. Am. Chem. Soc., 1989, 111, 8566 CrossRef CAS; J.-H. Lii and N. L. Allinger, J. Am. Chem. Soc., 1989, 111, 8576 CrossRef; N. L. Allinger, F. Li and L. Yan, J. Comput. Chem., 1990, 11, 848 CrossRef; N. L. Allinger, F. Li, L. Yan and J. C. Tai, J. Comput. Chem., 1990, 11, 868 CrossRef; N. L. Allinger, Y. H. Yuh and J.-H. Lii, J. Am. Chem. Soc., 1989, 111, 8551 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: The computational method and of the energetic and structural analysis. See DOI: 10.1039/c4ra11269f |
| This journal is © The Royal Society of Chemistry 2015 |