
Synthesis, molecular modeling, and biological evaluation of quinazoline derivatives containing the 1,3,4-oxadiazole scaffold as novel inhibitors of VEGFR2
Abstract
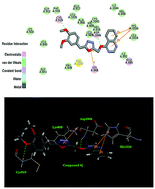
A series of 4-alkoxyquinazoline derivatives containing the 1,3,4-oxadiazole scaffold have been designed and synthesized, and their inhibitory activities were also tested against A549, MCF-7 and Hela. Of these compounds, 2-(3,4-dimethoxybenzyl)-5-((quinazolin-4-yloxy)methyl)-1,3,4-oxadiazole (compound 4j) showed the most potent inhibitory activity (IC50 = 0.23 μM for MCF-7, IC50 = 0.38 μM for A549 and IC50 = 0.32 μM for Hela) and the effect was better than the positive control drug Tivozanib (IC50 = 0.38 μM for MCF-7, IC50 = 0.62 μM for A549 and IC50 = 0.34 μM for Hela). A docking simulation was performed to position compound 4j into the VEGFR active site to determine the probable binding model. These results suggested that compound 4j with potent inhibitory activity in tumor growth inhibition may be a potential anticancer agent.
 Please wait while we load your content...
Please wait while we load your content...

