
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructure, antimicrobial activity, albumin- and DNA-binding of manganese(II)–sparfloxacinato complexes†
Marianthi
Zampakou
a,
Sofia
Balala
b,
Franc
Perdih
c,
Stavros
Kalogiannis
b,
Iztok
Turel
c and
George
Psomas
*a
aDepartment of General and Inorganic Chemistry, Faculty of Chemistry, Aristotle University of Thessaloniki, GR–54124 Thessaloniki, Greece. E-mail: gepsomas@chem.auth.gr; Fax: +30 2310997738; Tel: +30 2310997790
bDepartment of Nutrition and Dietetics, Faculty of Agriculture, Food Technology and Nutrition, Alexander Technological Educational Institution, Sindos, Thessaloniki, Greece
cFaculty of Chemistry and Chemical Technology, University of Ljubljana, Vecna pot 113, 1000 Ljubljana, Slovenia
First published on 8th January 2015
Abstract
Manganese(II) complexes with the quinolone antimicrobial agent sparfloxacin (Hsf) in the absence or presence of the nitrogen-donor heterocyclic ligands 1,10-phenanthroline (phen), 2,2′-bipyridine (bipy), 2,2′-bipyridylamine (bipyam) or pyridine (py) were synthesized and characterized with diverse physicochemical and spectroscopic techniques. The crystal structure of complex [Mn(sf)2(phen)]·4MeOH was determined by X-ray crystallography. In the resultant complexes, the deprotonated sparfloxacinato ligands are bidentately bound to manganese(II) through the pyridone oxygen and a carboxylato oxygen. The antimicrobial activity of the complexes was tested against four different microorganisms (Escherichia coli, Xanthomonas campestris, Staphylococcus aureus and Bacillus subtilis) and was found to be similar to or higher than free Hsf. The binding of the complexes to calf-thymus DNA (CT DNA) was monitored by UV spectroscopy and DNA viscosity measurements, which indicated intercalation as the most possible mode, and the DNA-binding constants of the complexes were calculated. The ability of the complexes to displace ethidium bromide (EB) from the EB–DNA complex was also investigated. Fluorescence emission spectroscopy was used to evaluate the interaction of the complexes with human or bovine serum albumin proteins revealing their binding with relatively high binding constant values.
Introduction
Manganese is among the most significant biometals mainly due to its presence in the active centre of many enzymes exhibiting diverse functionality.1,2 The anticancer agent SC-52608 and the MRI contrast agent Teslascan are the only manganese-containing compounds used in medicine up to now.3 Within the context of the attempt of bioinorganic chemists to develop novel metallodrugs, a lot of manganese compounds were tested for their in vitro bioactivity. The obtained preliminary results were quite interesting since some of the resultant manganese complexes exhibited noteworthy anticancer,4–6 antifungal7 and antimicrobial activity. The ligands of the manganese complexes showing antimicrobial activity present an interesting diversity; they may be drugs such as monensin A,8 thiopental sodium,9 chloramphenicol10 or enrofloxacin11 or ligands such as Schiff bases,12–15 derivatives of coumarin16,17 and 8-hydroxyquinoline,18 carbazone derivatives,19,20 carboxylic acids,21,22 hydrazides,23,24 and macrocycles.25,26Quinolones are a group of synthetic antibacterial agents used in the treatment of many infections.27,28 The main targets of quinolones are the participating in the DNA-replication enzymes gyrase (type II topoisomerases) and topoisomerase IV.29,30 Diverse metal complexes of quinolones have been reported exhibiting enhanced antiproliferative31–33 and antibacterial34,35 activity and increased binding affinity for DNA36–38 in comparison to the corresponding free quinolones.39 Sparfloxacin (Hsf)† (Fig. 1) is a third-generation quinolone, it is active against Gram-positive species such as Staphylococci and is used to treat chronic bronchitis and community-acquired pneumonia.28,29 The major advantages of Hsf are the good bioavailability and the long half-life which permit once-daily dosing, thus contributing to improved adherence to therapy and cost-effectiveness.40 In the literature, a Zn(II),41 two Ni(II)42,43 and three Cu(II)44,45 sparfloxacinato complexes have been structurally characterized and have exhibited enhanced biological behaviour in comparison to free Hsf.39
 | ||
| Fig. 1 Sparfloxacin (Hsf). | ||
Taking into consideration the wide-spread use of quinolones in medicine, the potential synergetic effects that occur in the presence of metals and as a continuation of our research,11,31–34,37–39,41–44 the interaction of manganese(II) with the third-generation quinolone antimicrobial drug sparfloxacin was investigated in the presence or absence of nitrogen-donor ligands such as 1,10-phenanthroline (phen), 2,2′-bipyridine (bipy), 2,2′-bipyridylamine (bipyam) or pyridine (py). The resultant complexes [Mn(sf)2(phen)]·4MeOH (1·4MeOH), [Mn(sf)2(bipy)] (2), [Mn(sf)2(bipyam)] (3), [Mn(sf)2(py)2] (4) and [Mn(sf)2(H2O)2] (5) were characterized with diverse physicochemical and spectroscopic techniques and the crystal structure of [Mn(sf)2(phen)]·4MeOH (1·4MeOH) was determined by X-ray crystallography. The biological impact of complexes 1–5 was evaluated in relevance to: (i) their antimicrobial activity by determining the minimum inhibitory concentration (MIC) against four microorganisms (i.e. Escherichia coli NCTC 29212 (E. coli), Xanthomonas campestris ATCC 1395 (X. campestris), Staphylococcus aureus ATCC 6538 (S. aureus) and Bacillus subtilis ATCC 6633 (B. subtilis)), (ii) their interaction with calf-thymus (CT) DNA investigated by UV spectroscopy and viscosity measurements in order to explore the binding mode and strength, (iii) their ability to displace the classical DNA–intercalator ethidium bromide (EB) from its CT DNA–EB complex as a means to investigate the existence of a potential intercalation monitored by fluorescence emission spectroscopy and (iv) their affinity to bovine (BSA) and human (HSA) serum albumins investigated by fluorescence spectroscopy, since the binding to these proteins which are involved in the transport of metal ions and compounds through the bloodstream may provide important information for the transportation of the compounds within the body, as a first means to potential applications.46
Experimental
Materials and instrumentation
Sparfloxacin, MnCl2·4H2O, bipy, phen, bipyam, py, KOH, NaCl, trisodium citrate, CT DNA, BSA, HSA and EB were purchased from Sigma-Aldrich Co and all solvents were purchased from Merck. All the chemicals and solvents were reagent grade and were used as purchased.DNA stock solution was prepared by dilution of CT DNA to buffer (containing 15 mM trisodium citrate and 150 mM NaCl at pH 7.0) followed by exhaustive stirring for three days, and kept at 4 °C for no longer than a week. The stock solution of CT DNA gave a ratio of UV absorbance at 260 and 280 nm (A260/A280) = 1.86, indicating that the DNA was sufficiently free of protein contamination.47 The concentration of the initial DNA solution per base pairs was determined by the UV absorbance at 260 nm after 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 dilution using ε = 6600 M−1 cm−1.48
:20 dilution using ε = 6600 M−1 cm−1.48
Infrared (IR) spectra (400–4000 cm−1) were recorded on a Nicolet FT-IR 6700 spectrometer with samples prepared as KBr disk. UV-visible (UV-vis) spectra were recorded as nujol mulls and in solution at concentrations in the range 10−5 to 10−3 M on a Hitachi U-2001 dual beam spectrophotometer. Room temperature magnetic measurements were carried out on a magnetic susceptibility balance of Sherwood Scientific (Cambridge, UK) by the Faraday method. C, H and N elemental analysis were performed on a Perkin-Elmer 240B elemental analyzer. Molar conductivity measurements of 1 mM DMSO solutions of the complexes were carried out with a Crison Basic 30 conductometer. Fluorescence spectra were recorded in solution on a Hitachi F-7000 fluorescence spectrophotometer. Viscosity experiments were carried out using an ALPHA L Fungilab rotational viscometer equipped with an 18 mL LCP spindle.
Synthesis of the complexes
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)pyridone, 1631 (very strong (vs)); νasym(CO2), 1604 (strong (s)); νsym(CO2), 1371 (s); Δ = νasym(CO2) − νsym(CO2) = 233 cm−1 (KBr disk); UV-vis: λ, nm (ε, M−1 cm−1) as nujol mull: 375 (shoulder (sh)), 306; in DMSO: 380 (400), 304 (10400). μeff = 5.97 BM. The complex is soluble in DMF and DMSO (ΛM = 10 μS cm−1, in 1 mM DMSO solution).
O)pyridone, 1631 (very strong (vs)); νasym(CO2), 1604 (strong (s)); νsym(CO2), 1371 (s); Δ = νasym(CO2) − νsym(CO2) = 233 cm−1 (KBr disk); UV-vis: λ, nm (ε, M−1 cm−1) as nujol mull: 375 (shoulder (sh)), 306; in DMSO: 380 (400), 304 (10400). μeff = 5.97 BM. The complex is soluble in DMF and DMSO (ΛM = 10 μS cm−1, in 1 mM DMSO solution).
[Mn(sf)2(bipy)], 2. A methanolic solution (5 mL) of bipy (0.2 mmol, 31 mg) was used instead of phen. A yellowish microcrystalline product [Mn(sf)2(bipy)], 2 (110 mg, yield: 55%) was collected after three days. Anal. calcd for C48H50F4MnN10O6 (MW = 993.92) C 58.01, H 5.07, N 14.09%; found: C 57.87, H 4.88, N 13.85%. IR: νmax, cm−1; ν(C
O)pyridone, 1632 (vs); νasym(CO2), 1603 (vs); νsym(CO2), 1379 (vs); Δ = 224 cm−1 (KBr disk); UV-vis: λ, nm (ε, M−1 cm−1) as nujol mull: 387 (sh), 307, 285; in DMSO: 382 (650), 305 (sh) (9500), 286 (14500). μeff = 6.01 BM. The complex is soluble in DMF and DMSO (ΛM = 14 μS cm−1, in 1 mM DMSO solution).
[Mn(sf)2(bipyam)], 3. A methanolic solution (5 mL) of bipyam (0.2 mmol, 39 mg) was used instead of phen. A pale-yellow microcrystalline product [Mn(sf)2(bipyam)], 3 (120 mg, yield: 60%) was deposited after a week. Anal. calcd for C48H51MnN11O6 (MW = 1008.94) C 57.14, H 5.10, N 15.27%; found: C 57.27, H 4.92, N 15.08%. IR: νmax, cm−1; ν(C
O)pyridone, 1634 (vs); νasym(CO2), 1598 (s); νsym(CO2), 1384 (s); Δ = 214 cm−1 (KBr disk); UV-vis: λ, nm (ε, M−1 cm−1) as nujol mull: 385 (sh), 309, 275; in DMSO: 380 (750), 308 (11500), 270 (15200). μeff = 5.95 BM. The complex is soluble in DMSO (ΛM = 8 μS cm−1, in 1 mM DMSO solution).
[Mn(sf)2(py)2], 4. 2 mL of py were added instead of phen. A pale-yellow microcrystalline product [Mn(sf)2(py)2], 4 (125 mg, yield: 63%) was deposited after twenty days. Anal. calcd for C48H52F4MnN10O6 (MW = 995.94) C 57.89, H 5.26, N 14.06%; found: C 57.71, H 5.12, N 13.91%. IR: νmax, cm−1; ν(C
O)pyridone, 1632 (vs); νasym(CO2), 1603 (s); νsym(CO2), 1383 (s); Δ = 220 cm−1 (KBr disk); UV-vis: λ, nm (ε, M−1 cm−1) as nujol mull: 385 (sh), 306, 277; in DMSO: 381 (750), 303 (12500), 280 (9700). μeff = 5.99 BM. The complex is soluble in DMF and DMSO (ΛM = 12 μS cm−1, in 1 mM DMSO solution).
O)pyridone, 1631 (vs); νasym(CO2), 1607 (s); νsym(CO2), 1380 (s); Δ = 227 cm−1 (KBr disk); UV-vis: λ, nm (ε, M−1 cm−1) as nujol mull: 380, 308 (sh), 288; in DMSO: 375 (250), 305 (sh) (3500), 290 (10500). μeff = 5.93 BM. The complex is soluble in DMF and DMSO (ΛM = 11 μS cm−1, in 1 mM DMSO solution).
X-ray structure determination
Single-crystal X-ray diffraction data were collected on Agilent Technologies SuperNova Dual diffractometer with an Atlas detector and Mo-Kα radiation (λ = 0.71073 Å) at room temperature. The data were processed using CrysAlis Pro.49 Structure was solved by direct methods implemented in SHELXS-97 and refined by a full-matrix least-squares procedure based on F2 with SHELXL-97.50 All the non-hydrogen atoms were refined anisotropically. Hydrogen atoms were readily located in difference Fourier maps and were subsequently treated as riding atoms in geometrically idealized positions, except in the NH group where they were refined freely with set isotropic temperature factors Uiso(H) = 1.2Ueq(N). Crystallographic data are listed in Table 1.| [Mn(sf)2(phen)]·4MeOH | |
|---|---|
| Empirical formula | C54H66F4MnN10O10 |
| M w | 1146.11 |
| T, K | 293(2) |
| Crystal system | Orthorhombic |
| Space group | Pccn |
| a (Å) | 14.9469(5) |
| b (Å) | 16.0111(5) |
| c (Å) | 23.5378(6) |
| α (°) | 90.00 |
| β (°) | 90.00 |
| γ (°) | 90.00 |
| V (Å3) | 5633.0(3) |
| Z | 4 |
| D calc (g cm−3) | 1.351 |
| μ (mm−1) | 0.313 |
| Data collected/unique/[I > 2σ(I)] | 21804/6465/3788 |
| Restraints/parameters | 0/366 |
| S | 1.024 |
| R 1, wR2 [I > 2σ(I)] | 0.0535/0.1180 |
| R 1, wR2 (all data) | 0.1068/0.1406 |
| Largest diff. peak/hole (e Å−3) | 0.310/−0.341 |
Determination of minimum inhibitory concentration
The antimicrobial activities of the compounds under investigation, Hsf and complexes 1–5, were evaluated by determining the MIC values towards four bacterial species, namely two Gram negative (E. coli NCTC 29212 and X. campestris ATCC 1395) and two Gram positive (S. aureus ATCC 6538 and B. subtilis ATCC 6633). Cultures of the microbial strains were grown on a rich selective agar medium and stored at 4 °C. The selective media used were nutrient agar or broth for B. subtilis and S. aureus, yeast mold agar or broth for X. campestris and Luria broth or agar for E. coli. Cells picked from the surface of the stored cultures were used to initiate liquid pre-cultures of the same selective medium at an initial turbidity of roughly 1 McFarland unit. Pre-cultures were incubated for 24 h in a rotary shaking incubator and subsequently they were used to inoculate the test cultures used for the determination of MIC at an initial turbidity of 0.5 McFarland units. The test cultures consisted of Mueller–Hinton broth (Deben Diagnostics Ltd) containing different concentrations of the compounds. Different concentrations were achieved as follows: the compounds were freshly dissolved in DMSO to a concentration of 5 mg mL−1 and they were diluted by a solution containing 5% v/v DMSO in sterile water, using the method of progressive double dilution. Thus working solutions with decreasing concentrations of the compounds under investigation were achieved. The working solutions were subsequently diluted to the final desired concentration by addition to the growth medium at a proportion of 2:98. MIC values were determined as the lowest concentrations of the tested compounds that inhibited visible growth of each respective organism after a 24 h incubation.51 All test cultures were grown in triplicates and for the determination of MIC, growth had to be inhibited in at least two cultures of the triplicate. Incubation temperature at all stages was 37 °C except for X. campestris that was cultivated at 28 °C.
DNA binding studies
 versus [DNA] using the UV spectra of the compound recorded for a constant concentration in the absence or presence of CT DNA for diverse r values. The Wolfe–Shimer equation is given below:
versus [DNA] using the UV spectra of the compound recorded for a constant concentration in the absence or presence of CT DNA for diverse r values. The Wolfe–Shimer equation is given below: | (1) |
 vs. [Q]. The Stern–Volmer equation is the following:
vs. [Q]. The Stern–Volmer equation is the following: | (2) |
Albumin binding experiments
The protein binding study was performed by tryptophan fluorescence quenching experiments using bovine (BSA, 3 μM) or human serum albumin (HSA, 3 μM) in buffer (containing 15 mM trisodium citrate and 150 mM NaCl at pH 7.0). The quenching of the emission intensity of tryptophan residues of BSA at 342 nm or HSA at 351 nm was monitored using complexes 1–5 as quencher with increasing concentration. The fluorescence spectra were recorded from 300 to 500 nm at an excitation wavelength of 295 nm.53 The sparfloxacinato complexes in buffer solutions did not exhibit under the same experimental conditions (i.e. excitation at 295 nm) any fluorescence emission. The influence and the extent of the inner-filter effect on the measurements was estimated with the following formula: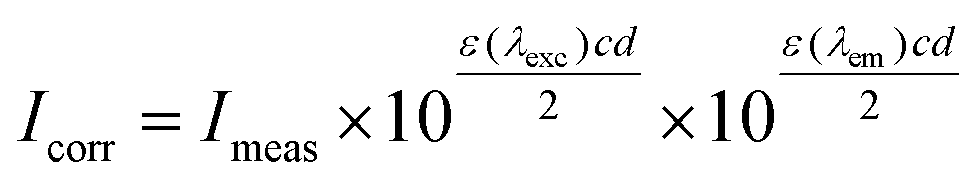 | (3) |
The Stern–Volmer and Scatchard equations (eqn (4)–(6))53 and graphs have been used in order to study the interaction of each quencher with serum albumins and calculate the dynamic quenching constant KSV (in M−1), the approximate quenching constant kq (in M−1 s−1), the association binding constant K (in M−1) and the number of binding sites per albumin n. According to the Stern–Volmer quenching equation:
 | (4) |
 vs. [Q]. From the equation:
vs. [Q]. From the equation:| KSV = kqτ0 | (5) |
 | (6) |
 versus
versus and n is given by the ratio of y intercept to the slope.
and n is given by the ratio of y intercept to the slope.
Results and discussion
Synthesis and characterization of the complexes
The complexes were synthesized via the reaction of sparfloxacin, initially deprotonated by KOH, with MnCl2·4H2O in the presence of the corresponding co-ligand (N,N′-donor = phen, bipy, bipyam for 1–3, respectively, py for 4 and H2O for 5), according to the following equations:| MnCl2·4H2O + 2Hsf + 2KOH + N,N′-donor → [Mn(sf)2(N,N′-donor)] + 2KCl + 6H2O | (7) |
| MnCl2·4H2O + 2Hsf + 2KOH + 2py → [Mn(sf)2(py)2] + 2KCl + 6H2O | (8) |
| MnCl2·4H2O + 2Hsf + 2KOH → [Mn(sf)2(H2O)2] + 2KCl + 4H2O | (9) |
The resultant complexes 1–5 are soluble in DMSO and are not electrolytes; the values of the molar conductivity (ΛM) of 1 mM DMSO solution of the complexes are in the range 8–14 μS cm−1 and indicate their non-dissociation in DMSO solution. The complexes were characterized by elemental analysis, IR and UV-vis spectroscopic techniques and magnetic measurements at room temperature. Additionally, the structure of 1 was determined by X-ray crystallography.
The deprotonation and the binding mode of sparfloxacin in complexes 1–5 were confirmed by IR spectroscopy. In the IR spectra of complexes 1–5, the band attributed to the ν(CO)pyridone stretching vibration shifted from 1640 cm−1 in free Hsf to 1631–1634 cm−1 suggesting coordination of the sparfloxacinato ligands to manganese via the pyridone oxygen. Additionally, the absence of the ν(O–H) stretching vibration of free Hsf at 3460 (broad, medium) cm−1 was indicative of the deprotonation of its carboxylate group upon binding to metal ion. The ν(CO)carboxyl and ν(C–O)carboxyl stretching vibrations of the carboxyl group (–COOH) of free Hsf at 1715 (vs) cm−1 and 1292 (s) cm−1, respectively, exhibited in the spectra of the complexes a shift in the range 1598–1607 cm−1 and 1371–1384 cm−1; these bands may now be characterized as antisymmetric, νasym(CO2), and symmetric, νsym(CO2), stretching vibrations of the carboxylato group, respectively. According to the Δ values [Δ = νasym(CO2) − νsym(CO2)] which appear in the range 214–233 cm−1, a monodentate coordination mode of the carboxylate group of sparfloxacinato ligands is suggested.55 Thus, the deprotonated sparfloxacinato ligands are coordinated to manganese in a bidentate mode via the pyridone oxygen and a carboxylato oxygen forming a six-membered chelate ring.41–44
The UV-vis spectra of complexes 1–5 were recorded in DMSO solution and as nujol mull and were similar showing that the compounds have the same structure in solution. The band at 375–382 nm (ε = 250–750 M−1 cm−1) can be attributed to a charge transfer transition while the bands found in the UV region are assigned to intra-ligand transitions of the complexes. The stability and integrity of the compounds in solution may be concluded from: (i) the same UV-vis spectral features presented in solid state (nujol), in solution (DMSO) and in the presence of the buffer solution used in the biological experiments (150 mM NaCl, 15 mM trisodium citrate, pH = 7.0), (ii) the molar conductivity measurements (8–14 μS cm−1 in 0.1 mM in DMSO) which suggest non-dissociation, (iii) the formation of multiple chelate rings as discussed below in the crystal structure description, and (iv) the comparison with previously reported metal–quinolone complexes.11,38,39,41–44,56
The observed values of μeff (=5.93–6.01 BM) for the complexes are close to the spin-only value (=5.92 MB) at room temperature and typical for mononuclear high-spin Mn(II) complexes with d5 configuration (S = 5/2).57
Structure of the complexes
 | ||
| Fig. 2 A drawing of the molecular structure of 1 with only the heteroatoms labelling. Hydrogen atoms and methanol solvate molecules are omitted for clarity. | ||
| Bond | Distance (Å) | Bond | Distance (Å) |
|---|---|---|---|
| Mn(1)–O(1) | 2.1190(18) | O(1)–C(1) | 1.258(3) |
| Mn(1)–O(3) | 2.1381(17) | O(2)–C(1) | 1.254(3) |
| Mn(1)–N(1) | 2.276(2) | O(3)–C(4) | 1.264(3) |
| Bond angle | (°) | Bond angle | (°) |
|---|---|---|---|
| O(1)–Mn(1)–O(1′) | 162.09(10) | O(3)–Mn(1)–O(3′) | 95.44(10) |
| O(1)–Mn(1)–O(3) | 81.90(7) | O(3)–Mn(1)–N(1) | 96.12(7) |
| O(1)–Mn(1)–O(3′) | 86.07(7) | O(3)–Mn(1)–N(1′) | 168.42(8) |
| O(1)–Mn(1)–N(1) | 94.79(8) | N(1)–Mn(1)–N(1′) | 72.34(11) |
| O(1)–Mn(1)–N(1′) | 99.66(7) |
The Mn(II) atom, Mn(1), is six-coordinated exhibiting a distorted octahedral geometry with a MnO4N2 chromophore consisted of four oxygen atoms from two sparfloxacinato ligands and two nitrogen atoms from the bidentate phen ligand occupying the six vertices of octahedron. The Mn–Ocarb distances [Mn(1)–O(1) = 2.1190(18) Å] are close to Mn–Opyr distances [Mn(1)–O(3) = 2.1381(17) Å] and shorter than the Mn–N distances [Mn(1)–N(1) = 2.276(2) Å]. The two pyridone oxygen atoms [O(3)–Mn(1)–O(3′) = 95.44(10)°] are cis to each other and the two carboxylato oxygen atoms [O(1)–Mn(1)–O(1′) = 162.09(10)°] are in a trans arrangement. Similar arrangement of the coordinated oxygen atoms of the sparfloxacinato ligands around the metal ion (i.e. cis pyridone and trans carboxylato oxygen atoms)39 has been also observed in the crystal structures of the complexes [Ni(sf)2(py)2] (O(2)–Ni(1)–O(21) = 89.05(6)° and O(1)–Ni–O(22) = 174.54(6)°),42 [Ni(sf)2(phen)] (O(1)–Ni(1)–O(1)′ = 90.78(8)° and O(2)–Ni–O(2)′ = 172.26(7)°),43 and [Zn(sf)2(phen)] (O(28)–Zn(1)–O(28)′ = 94.00(12)° and O(27)–Zn(1)–O(27)′ = 169.90(11)°).41
The overall arrangement of the two sparfloxacinato ligands in the molecule could be considered as a pincer trapping the phen ligand of a neighbouring molecule creating a pair of host–guest molecules and a chain is formed (Fig. S1†), as it was previously observed and thoroughly described in the crystal structure of [Zn(sf)2(phen)].41 The host–guest structure is stabilized by the formation of a hydrogen bond between the piperazine nitrogen atom of the host molecule (donor) and the unbonded carboxylic oxygen of the sparfloxacinato ligand in the guest molecule (acceptor) (Fig. S2†). This structure is further stabilized by weak π⋯π [3.9982(16) Å] and CH⋯π interactions. The Mn(phen) plane of the guest molecule creates a torsion angle of about 103° with the Mn(phen) plane of the host molecule. In the crystal lattice there are four methanol solvate molecules. Two of them are hydrogen bonded to the piperazine nitrogen atoms and additional two methanol molecules act as hydrogen bond donors to carboxylic groups and as acceptors to amino moieties of the sparfloxacinato ligands (Table S1, Fig. S2†).
The phen ligand is almost planar with the manganese atom lying in this plane. The Mn–N bond distances [Mn(1)–N(1) = 2.276(2) Å] and the N(1)–Mn(1)–N(1′) angle [72.34(11)°] are within the range of reported values of other chelating polycyclic α-diimines.11,58,59
Proposed structures for complexes 2–5
Based on the experimental data (IR spectroscopy, elemental analysis, molar conductivity and magnetic measurements at ambient temperature) and after a comparison to the recent literature, we may propose a structure for complexes 2–5. Complexes [Mn(sf)2(bipy)], 2 and [Mn(sf)2(bipyam)], 3 are expected to have similar structure to that of complex 1, while for complexes 4 and 5 a structure similar to that of [Ni(sf)2(py)2],42 [Co(erx)2(py)2] (Herx = enrofloxacin),60 [Zn(erx)2(py)2],61 and [Zn(levo)2(H2O)2] (Hlevo = levofloxacin)41 is anticipated, as described below. According to the IR spectra, the sparfloxacinato ligands are deprotonated being in bidentate binding mode coordinated to manganese ion via the pyridone and a carboxylato oxygen. On the basis of the magnetic data at room temperature, the complexes are mononuclear exhibiting an octahedral geometry around Mn(II) ion. The octahedron is formed by four oxygen atoms of the two sparfloxacinato ligands and two nitrogen or oxygen atoms from the pyridine or aqua ligands in complex 4 or 5, respectively.Antimicrobial activity of the complexes
The antimicrobial activity of Hsf and its manganese(II) complexes 1–5 were evaluated by monitoring the growth of two Gram-positive (B. subtilis, S. aureus) and two Gram-negative (X. campestris, E. coli) bacterial strains in the presence of various concentrations of the complexes ranging from 0 to 32 μg mL−1; the obtained minimum inhibitory concentration (MIC) values are presented in Table 3.| Compound | B. subtilis | S. aureus | X. campestris | E. coli |
|---|---|---|---|---|
| Hsf | 0.500 | 0.250 | 0.500 | 0.0625 |
| [Mn(sf)2(phen)], 1 | 0.500 | 0.500 | 0.250 | 0.0625 |
| [Mn(sf)2(bipy)], 2 | 0.250 | 0.500 | 0.500 | 0.0625 |
| [Mn(sf)2(bipyam)], 3 | 1.000 | 2.000 | 1.000 | 0.125 |
| [Mn(sf)2(py)2], 4 | 0.250 | 0.500 | 0.500 | 0.0625 |
| [Mn(sf)2(H2O)2], 5 | 0.250 | 0.500 | 0.500 | 0.0625 |
The results suggest that Hsf and complexes 1–5 are active against both Gram-positive and Gram-negative bacteria with MIC values being in the range 0.0625–1.000 μg mL−1. In most cases the complexes exhibited similar or higher antimicrobial activity than free Hsf. Especially, complexes 2, 4 and 5 were more active than Hsf against B. subtilis, as well as complex 1 against X. campestris. On the other hand, [Mn(sf)2(bipyam)] 3 was less active than Hsf against all bacteria tested. The lowest MIC values presented by all compounds were against E. coli.
The reported Mn(II)–quinolone complexes are rather limited up to date; thus, we cannot firmly propose the main factors that affect the antimicrobial activity according to the literature.11,27,39,62 As a first approach, among the five universally accepted factors (chelate effect of ligands, nature of ligands, nuclearity, total charge, existence and nature of counter-ions)63 responsible for the antimicrobial activity of the complexes, the chelate effect and the nature of the ligands (sparfloxacinato and N-donor ligands) may be the prevailing factors present in complexes 1–5 that contribute to improved antimicrobial activity.
Interaction of the complexes with CT DNA
The activity of quinolone compounds as potent antibacterial agents is mainly focused on the inhibition of DNA-replication by targeting essential type II bacterial topoisomerases such as DNA gyrase and topoisomerase IV;27–29,64 thus, the evaluation of the interaction of quinolones and their complexes with DNA is of great significance. In general, metal complexes bind to double-stranded DNA via covalent (replacement of a labile ligand of the complex by a nitrogen base of DNA) or noncovalent interactions, including intercalation inside the DNA helix (via π → π* stacking interaction of the complex and DNA nucleobases), groove binding along major or minor groove of the DNA helix (presence of van der Waals interaction or hydrogen-bonding or hydrophobic bonding) and external/electrostatic interactions (development of Coulomb forces between metal complexes and the phosphate groups being on the surface of DNA).39,65,66Study of the DNA-binding using UV spectroscopy
UV spectroscopy is a technique that provides useful information concerning the interaction mode and the strength of binding of complexes with DNA. In this context, the UV spectra either of a CT DNA solution (C ∼ 0.75–1.2 × 10−4 M) were recorded in the presence of the complexes 1–5 at increasing amounts (for different [complex]/[DNA] mixing ratios (=r)) or of a DMSO solution of the complexes 1–5 (1–2 × 10−5 M) in the presence of CT DNA at diverse r values, since any interaction between the complex and CT DNA may perturb the band of CT DNA at 258–260 nm or the intra-ligand transition bands of the complexes, respectively, during the titrations.41–43The UV spectra of a CT DNA solution in the presence of the complex 3 at increasing r values are shown representatively in Fig. 3(A). Quite similar is the behaviour of CT DNA in the presence of the other complexes. The slight decrease of the intensity at λmax = 258 nm indicates that the interaction of CT DNA with the complex takes place resulting in the formation of a new complex with double-helical CT DNA.
 | ||
| Fig. 3 (A) UV spectra of CT DNA (7.5 × 10−5 M) in buffer solution (150 mM NaCl and 15 mM trisodium citrate at pH 7.0) in the absence or presence of [Mn(sf)2(bipyam)], 3. The arrow shows the changes upon increasing amounts of the complex (up to r = 0–0.6). (B) UV spectra of DMSO solution (1 × 10−5 M) of [Mn(sf)2(phen)], 1 in the presence of increasing amounts of CT DNA. The arrows show the changes upon increasing amounts of CT DNA (r′ = 0–0.8). | ||
In the UV spectra of complex 1 (1 × 10−5 M) upon the addition of increasing amounts of CT DNA (up to r′ = [DNA]/[complex] = 1.6), the intra-ligand band at 306 nm (band I) shows a hypochromism up to 29% with a slight hypsochromism (Fig. 3(B) for (r′ = 0–0.8)). Quite similar is the behaviour of complexes 2–5 upon the addition of CT DNA with the hypochromism being in the range 6–40% (Table 4). In general, the observed hypochromism may be attributed to π → π stacking interaction between the aromatic chromophore (from sparfloxacinato and/or the N-donor ligands) of the complex and DNA base pairs consistent with the intercalative binding mode. The existing results suggest that the complexes can bind to CT DNA, possibly by intercalation, although the exact mode of DNA-binding cannot be concluded only by UV spectroscopic titration studies; nevertheless, more experiments are necessary in order to further clarify the binding mode and possible verify the existence of intercalation.67
| Band (λ, nm) (ΔA (%), Δλ (nm)) | K b (M−1) | |
|---|---|---|
| Hsf43 | 308 (−12, −12) | 1.71(±0.02) × 105 |
| [Mn(sf)2(phen)], 1 | 306 (−29, −3) | 1.01(±0.13) × 106 |
| [Mn(sf)2(bipy)], 2 | 304 (sh) (−8, 0) | 1.15(±0.20) × 106 |
| [Mn(sf)2(bipyam)], 3 | 309 (−40, 0) | 3.84(±0.22) × 105 |
| [Mn(sf)2(py)2], 4 | 303 (−10, +5) | 1.25(±0.05) × 105 |
| [Mn(sf)2(H2O)2], 5 | 308 (sh) (−6, 0) | 3.42(±0.14) × 105 |
The values of the DNA-binding constant (Kb) of complexes 1–5 were calculated using Wolfe–Shimer equation (eqn (1))52 and plots  versus [DNA] (Fig. S3†) and are presented in Table 4. The Kb values are high and most of them are higher than that of free Hsf suggesting a significant increase of the Kb value when coordinated to Mn(II). The Kb values indicate a strong binding of the complexes to CT DNA, with complexes 1 and 2 having the highest Kb values (=1.01(±0.13) × 106 M−1 and 1.15(±0.20) × 106 M−1, respectively) among the compounds, which are higher than that of the classical intercalator EB (=1.23(±0.07) × 105 M−1) as calculated in ref. 68. The Kb values of complexes 1 and 2 are among the highest of values of the metal–quinolone complexes reported so far.11,39,41–43,60,61
versus [DNA] (Fig. S3†) and are presented in Table 4. The Kb values are high and most of them are higher than that of free Hsf suggesting a significant increase of the Kb value when coordinated to Mn(II). The Kb values indicate a strong binding of the complexes to CT DNA, with complexes 1 and 2 having the highest Kb values (=1.01(±0.13) × 106 M−1 and 1.15(±0.20) × 106 M−1, respectively) among the compounds, which are higher than that of the classical intercalator EB (=1.23(±0.07) × 105 M−1) as calculated in ref. 68. The Kb values of complexes 1 and 2 are among the highest of values of the metal–quinolone complexes reported so far.11,39,41–43,60,61
DNA-binding study with viscosity measurements
The measurement of the viscosity of a DNA solution upon addition of the complexes is a method that provides useful information in the attempt to clarify the DNA-binding mode, since the DNA-viscosity is quite sensitive to DNA-length changes; this relation is given by the equation L/L0 = (η/η0)1/3, with η/η0 denoting the relative solution viscosity and L/L0 the relative DNA length.65 In many cases, the DNA-viscosity measurements may be used complimentary to the UV spectroscopic titration studies.In the present case, the viscosity measurements were carried out on CT DNA solutions (0.1 mM) upon addition of increasing amounts of complexes 1–5 (up to the value of r = 0.35) (Fig. 4). The addition of the complexes into a DNA solution resulted in a significant increase of the relative viscosity of DNA; this increase may be explained by an increase of the overall DNA length caused by the insertion of the complexes in between the DNA base pairs which increases the separation of base pairs at intercalation sites.41–43,65 The observed increase of the DNA viscosity in the presence of compounds is evidence of an intercalative binding mode to DNA; a conclusion which clarifies and enforces the preliminary findings from UV spectroscopic studies showing a hypochromism which can now be considered evidence of intercalation.65
 | ||
| Fig. 4 Relative viscosity (η/η0)1/3 of CT DNA ([DNA] = 0.1 mM) in buffer solution (150 mM NaCl and 15 mM trisodium citrate at pH 7.0) in the presence of complexes 1–5 at increasing amounts (r = [complex]/[DNA]). | ||
Competitive study with ethidium bromide
Ethidium bromide (EB = 3,8-diamino-5-ethyl-6-phenyl-phenanthridinium bromide) intercalates to DNA through the planar EB phenanthridine ring. The result of the intercalation is the appearance of an intense fluorescence emission band at 592 nm, when excited at 540 nm, due to the formation of the EB–DNA compound. Thus, EB is considered a typical indicator of intercalation since a quenching of the DNA-induced EB fluorescence emission may appear when an equal or stronger intercalating compound is added into a solution of the EB–DNA compound.69 Complexes 1–5 do not show any significant fluorescence emission at room temperature in solution or in the presence of CT DNA under the same experimental conditions and their addition to a solution containing EB does not provoke quenching of free EB fluorescence and new peaks do not appear in the spectra. Therefore, the changes observed in the fluorescence emission spectra of a solution containing the EB–DNA compound upon addition of the complexes can be used to investigate the ability of the complexes to displace EB from the EB–DNA complex.The fluorescence emission spectra of pre-treated EB–DNA ([EB] = 20 μM, [DNA] = 26 μM) were recorded for increasing amounts of the complexes up to the value of r = 0.07 (for 1 in Fig. 5(A)). The addition of complexes 1–5 at diverse r values resulted in a moderate to significant decrease of the intensity of the emission band of the DNA–EB system at 592 nm (the final fluorescence is up to 19–51% of the initial EB–DNA fluorescence intensity in the presence of the complexes, Table 5) indicating a moderate to significant competition of the complexes with EB in binding to DNA (Fig. 5(B)). The observed quenching of DNA–EB fluorescence by the complexes suggests that the complexes can displace EB from the DNA–EB compound, thus revealing the interaction with CT DNA by the intercalative mode.11,39,41–43,61,65
 | ||
| Fig. 5 (A) Emission spectra (λexit = 540 nm) for EB–DNA ([EB] = 20 μM, [DNA] = 26 μM) in buffer solution in the absence and presence of increasing amounts of complex 1 (up to the value of r = 0.065). The arrow shows the changes of intensity upon increasing amounts of 1. (B) Plot of EB relative fluorescence intensity at λem = 592 nm (%) vs. r (r = [complex]/[DNA]) (150 mM NaCl and 15 mM trisodium citrate at pH = 7.0) in the presence of complexes 1–5 (up to 23% of the initial EB–DNA fluorescence intensity for 1, 37% for 2, 19% for 3, 37% for 4 and 51% for 5). | ||
| Compound | EB–DNA fluorescence quenching, (ΔI/Io) (%) | K SV (M−1) |
|---|---|---|
| Hsf43 | 80% | 2.13(±0.09) × 106 |
| [Mn(sf)2(phen)], 1 | 77% | 1.54(±0.06) × 106 |
| [Mn(sf)2(bipy)], 2 | 63% | 5.13(±0.21) × 105 |
| [Mn(sf)2(bipyam)], 3 | 81% | 2.12(±0.06) × 106 |
| [Mn(sf)2(py)2], 4 | 63% | 6.61(±0.17) × 105 |
| [Mn(sf)2(H2O)2], 5 | 49% | 2.85(±0.14) × 105 |
The Stern–Volmer plots of DNA–EB for complexes 1–5 (Fig. S4†) illustrate that the quenching of EB–DNA by the complexes is in good agreement (R = 0.99) with the linear Stern–Volmer equation (eqn (2)) proving that the replacement of EB from EB–DNA by each complex results in a decrease in the fluorescence intensity.41–43,65 The obtained values of KSV (Table 5) are relatively high showing the tight binding of the complexes to DNA with complex 3 exhibiting the highest Ksv value (=2.12(±0.06) × 106 M−1) among the complexes. The KSV values of complexes 1–5 are of the same magnitude to those of a series of metal complexes with quinolones as ligands.39
Interaction of the complexes with serum albumins
Serum albumin (SA) is a protein in plasma responsible for the transport of ions and drugs through the bloodstream to cells and tissues. Thus, the interaction of potential drugs with SA as a first means of their transport towards their targets in the body is worth studying for future medical use.46,70 Human serum albumin (HSA) and its homologue bovine serum albumin (BSA) were used to study the interaction with complexes 1–5 from tryptophan emission-quenching experiments. HSA has a tryptophan (Trp-214) and BSA has two tryptophans (Trp-134 and Trp-212); their solutions exhibit an intense fluorescence emission when excited at 295 nm, with λem,max = 351 nm and 343 nm, respectively. The sparfloxacinato complexes 1–5 did not show any significant fluorescence emission under the same experimental conditions (i.e. λexc = 295 nm).41 The inner-filter effect was calculated with eqn (3) and slightly affected the measurements.54The addition of complexes 1–5 to a HSA solution results in relatively significant fluorescence quenching, as calculated after the correction of the initial fluorescence spectra, (up to ∼85% for 5, Fig. 6(A)). The quenching of BSA fluorescence upon addition of complexes 1–5 is much more pronounced up to ∼92% for complexes 1 and 2 (Fig. 6(B)). This quenching may be attributed to possible changes in protein secondary structure of SA indicating the binding of the compounds to SA.70
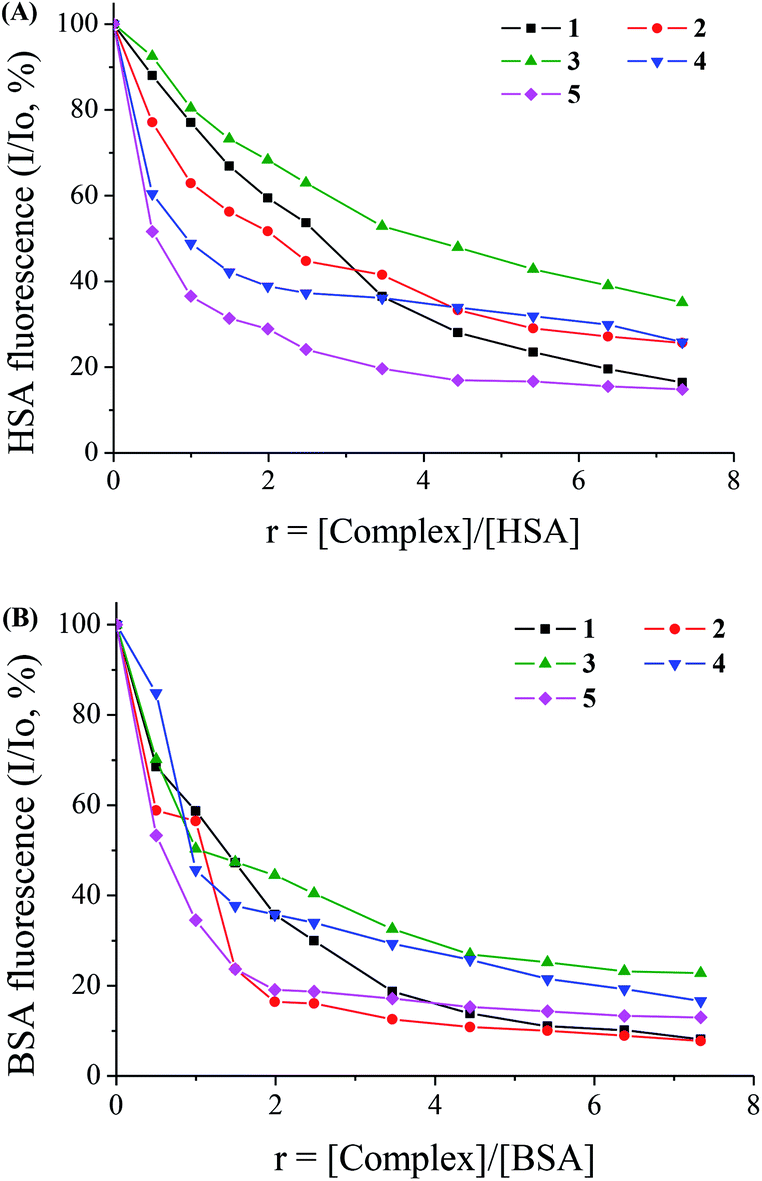 | ||
| Fig. 6 (A) Plot of relative fluorescence intensity at λem = 351 nm (%) vs. r (r = [complex]/[HSA]) for complexes 1–5 (up to 16.5% of the initial HSA fluorescence for 1, 25.5% for 2, 35% for 3, 25.5% for 4 and 15% for 5) in buffer solution (150 mM NaCl and 15 mM trisodium citrate at pH 7.0). (B) Plot of relative fluorescence intensity at λem = 343 nm (%) vs. r (r = [complex]/[BSA]) for complexes 1–5 (up to 8% of the initial BSA fluorescence for 1 and for 2, 22% for 3, 17% for 4 and 13% for 5) in buffer solution (150 mM NaCl and 15 mM trisodium citrate at pH 7.0). | ||
The quenching constant values (kq) for the interaction of complexes 1–5 with the albumins were calculated using the Stern–Volmer quenching equation (eqn (4)) and the corresponding Stern–Volmer plots (Fig. S5 and S6†) and are given in Table 6. The kq values (>1012 M−1 s−1) are higher than diverse kinds of quenchers for biopolymers fluorescence (2.0 × 1010 M−1 s−1) indicating the existence of static quenching mechanism.71 These values indicate good SA quenching ability and the kq values of the complexes are similar or higher, in most cases, than the corresponding values of free Hsf, with 5 exhibiting the highest quenching ability for HSA (kq(HSA),5 = 3.53(±0.20) × 1013 M−1 s−1) and complexes 1 and 2 for BSA (kq(BSA),1 = 5.26(±0.23) × 1013 M−1 s−1 and kq(BSA),2 = 5.30(±0.23) × 1013 M−1 s−1). The values of the quenching constants are within the range found for a series of metal–quinolone complexes.39
| Complex | K sv (M−1) | k q (M−1 s−1) | K (M−1) | n |
|---|---|---|---|---|
| HSA | ||||
| Hsf41 | 1.26(±0.04) × 105 | 1.26(±0.04) × 1013 | 6.04(±0.31) × 104 | 0.65 |
| [Mn(sf)2(phen)], 1 | 2.35(±0.13) × 105 | 2.35(±0.13) × 1013 | 5.66(±0.35) × 104 | 1.58 |
| [Mn(sf)2(bipy)], 2 | 1.33(±0.05) × 105 | 1.33(±0.05) × 1013 | 2.27(±0.11) × 105 | 0.89 |
| [Mn(sf)2(bipyam)], 3 | 8.39(±0.10) × 104 | 8.39(±0.10) × 1012 | 7.76(±0.30) × 104 | 1.03 |
| [Mn(sf)2(py)2], 4 | 1.16(±0.11) × 105 | 1.16(±0.11) × 1013 | 7.67(±0.24) × 105 | 0.74 |
| [Mn(sf)2(H2O)2], 5 | 3.53(±0.20) × 105 | 3.53(±0.20) × 1013 | 7.59(±0.30) × 105 | 0.90 |
| BSA | ||||
| Hsf41 | 1.04(±0.05) × 105 | 1.04(±0.05) × 105 | 6.30(±0.25) × 104 | 1.40 |
| [Mn(sf)2(phen)], 1 | 5.26(±0.23) × 105 | 5.26(±0.23) × 1013 | 2.66(±0.13) × 105 | 1.08 |
| [Mn(sf)2(bipy)], 2 | 5.30(±0.29) × 105 | 5.30(±0.23) × 1013 | 4.42(±0.31) × 105 | 1.04 |
| [Mn(sf)2(bipyam)], 3 | 1.73(±0.08) × 105 | 1.73(±0.08) × 1013 | 3.31(±0.16) × 105 | 0.88 |
| [Mn(sf)2(py)2], 4 | 2.03(±0.11) × 105 | 2.03(±0.11) × 1013 | 3.40(±0.40) × 105 | 0.93 |
| [Mn(sf)2(H2O)2], 5 | 4.83(±0.32) × 105 | 4.83(±0.32) × 1013 | 6.83(±0.04) × 105 | 0.94 |
The values of the association binding constant (K) of the complexes to the SAs were calculated using the Scatchard equation (eqn (6)) and the corresponding Scatchard plots (Fig. S7 and S8†) and are given in Table 6. The K values of complexes 1–5 are relatively high and higher than that of free Hsf with complexes 4 and 5 having the highest K value for HSA (K(HSA),4 = 7.67(±0.24) × 105 M−1, K(HSA),5 = 7.59(±0.30) × 105 M−1), while in the case of BSA complex 5 exhibits the highest K value among the compounds (K = 6.83(±0.04) × 105 M−1). Comparing the affinity of the complexes for BSA and HSA (K values), it is obvious that all complexes except 4 show higher affinity for BSA than HSA. In general, the K values of the complexes are of the same magnitude with those calculated for a series of metal–quinolone complexes.39
In general, the values of the binding constants (K) of the complexes to SA are in the range 5.66(±0.35) × 104 to 7.67(±0.24) × 105 M−1 which could be considered rather optimum suggesting their binding to SAs and possible transfer. All the K values are significantly lower than the value of the association constant of the protein avidin with diverse compounds (K ≈ 1015 M−1), an interaction which is considered the strongest known non-covalent one;72,73 indeed, the binding constants are reasonably low so that the complexes can get reversibly released when arriving at their targets.
Conclusions
The synthesis and characterization of the mononuclear manganese(II) complexes with the third-generation quinolone antibacterial agent sparfloxacin in the absence or presence of the nitrogen-donor heterocyclic ligands 1,10-phenanthroline, 2,2′-bipyridine, 2,2′-bipyridylamine or pyridine was successfully achieved. In the resultant complexes, sparfloxacinato ligands are deprotonated being bidentately coordinated to manganese via the pyridone and a carboxylato oxygen. The crystal structure of the complex [Mn(sf)2(phen)] was determined by X-ray crystallography revealing a distorted octahedral geometry for Mn(II) presenting a similar arrangement of the atoms around manganese with previously reported Zn–sparfloxacinato complexes.UV spectroscopy studies and viscosity measurements were employed to investigate the interaction of the complexes with CT DNA. Complexes [Mn(sf)2(phen)], 1 and [Mn(sf)2(bipy)], 2, exhibit the highest DNA binding constants, Kb (Kb(1) = 1.01(±0.13) × 106 M−1 and Kb(2) = 1.15(±0.20) × 106 M−1, respectively), among the complexes examined, which are among the highest values of the reported metal–quinolone complexes39 and higher than the Kb value of EB as calculated in our lab.68 Competitive binding studies with EB revealed the significant EB-displacing ability of the complexes from the EB–CT DNA complex indicating indirectly the intercalation as the most possible interaction mode to CT DNA, a conclusion which is in accordance to the viscometry measurements.
The antimicrobial activity of the complexes was estimated by MIC determination; in most cases and with the exception of complex 3, the complexes show similar or slightly better activity than free Hsf against the bacteria tested.
The interaction of all complexes with bovine or human serum albumins was studied using fluorescence emission spectroscopy. The complexes bind to both albumins BSA and HSA and have relatively high binding constants, K; the K values are in the range 5.66 × 104 to 7.67 × 105 M−1 and are indicative of the complexes' ability to bind to the albumin in order to get transferred towards their targets where they can get released since the K values are not too high.
In conclusion, the existing biological studies concerning the Mn(II)–sparfloxacinato complexes 1–5 are promising in regard to their potential application as antimicrobial agents; although the antibacterial activity of the complexes is similar or slightly higher than free Hsf, it is significant since the MIC values are very low.
Abbreviations
| B. subtilis | Bacillus subtilis |
| bipy | 2,2′-Bipyridine |
| bipyam | 2,2′-Bipyridylamine |
| BSA | Bovine serum albumin |
| CT | Calf-thymus |
| DMF | N,N-Dimethylformamide |
| DMSO | Dimethylsulfoxide |
| E. coli | Escherichia coli |
| EB | Ethidium bromide = 3,8-diamino-5-ethyl-6-phenyl-phenanthridinium bromide |
| Herx | Enrofloxacin |
| Hlevo | Levofloxacin |
| Hsf | Sparfloxacin = 5-amino-1-cyclopropyl-7-(cis-3,5-dimethyl-1-piperazinyl)-6,8-difluoro-1,4-dihydro-4-oxo-3-quinolinecarboxylic acid |
| HSA | Human serum albumin |
| MIC | Minimum inhibitory concentration |
| phen | 1,10-Phenanthroline |
| py | Pyridine |
| r | [Compound]/[CT DNA] mixing ratios |
| r′ | [DNA]/[complex] mixing ratios |
| s | Strong |
| S. aureus | Staphylococcus aureus |
| SA | Serum albumin |
| sf | Sparfloxacinato anion |
| sh | Shoulder |
| vs | Very strong |
| X. campestris | Xanthomonas campestris |
| Δ | ν asym(CO2) − νsym(CO2) |
Acknowledgements
This research has been co-financed by European Social Fund (ESF) and Greek national funds (National Strategic Reference Framework (NSRF): Archimides III. This project was supported by EU COST Action CM1105, the Slovenian Research Agency (ARRS) through project P1-0175 and partially by the infrastructure of the EN–FIST, Center of Excellence, Ljubljana, Slovenia.References
- E. J. Larson and V. L. Pecoraro, in Manganese Enzymes, ed. V. L. Pecoraro, VCH Publishers Inc, New York, 1992, pp. 1–28 Search PubMed.
- C. S. Mullins and V. L. Pecoraro, Coord. Chem. Rev., 2008, 252, 416–443 CrossRef CAS PubMed.
- Z. Guo and P. J. Sadler, Angew. Chem., Int. Ed., 1999, 38, 1512–1531 CrossRef CAS.
- M. Li, C. Chen, D. Zhang, J. Niu and B. Ji, Eur. J. Med. Chem., 2010, 45, 3169–3177 CrossRef CAS PubMed.
- D. Zhou, Q. Chen, Y. Qi, H. Fu, Z. Li, K. Zhao and J. Gao, Inorg. Chem., 2011, 50, 6929–6937 CrossRef CAS PubMed.
- X. Chen, L. Tang, Y. Sun, P. Qiu and G. Liang, J. Inorg. Biochem., 2010, 104, 1141–1147 CrossRef PubMed.
- D. P. Singh, K. Kumar and C. Sharma, Eur. J. Med. Chem., 2010, 45, 1230–1236 CrossRef CAS PubMed.
- P. Dorkov, I. N. Pantcheva, W. S. Sheldrick, H. Mayer-Figge, R. Petrova and M. Mitewa, J. Inorg. Biochem., 2008, 102, 26–32 CrossRef CAS PubMed.
- M. S. Refat, J. Mol. Struct., 2013, 1037, 170–185 CrossRef CAS PubMed.
- M. G. A. El-Wahed, M. S. Refat and S. M. El-Megharbel, J. Mol. Struct., 2008, 892, 402–413 CrossRef CAS PubMed.
- M. Zampakou, M. Akrivou, E. G. Andreadou, C. P. Raptopoulou, V. Psycharis, A. A. Pantazaki and G. Psomas, J. Inorg. Biochem., 2013, 121, 88–99 CrossRef CAS PubMed.
- S. Mandal, A. Rout, A. Ghosh, G. Pilet and D. Bandyopadhyay, Polyhedron, 2009, 28, 3858–3862 CrossRef CAS PubMed.
- G. Kumar, S. Devi, R. Johari and D. Kumar, Eur. J. Med. Chem., 2012, 52, 269–274 CrossRef CAS PubMed.
- M. Patil, R. Hunoor and K. Gudasi, Eur. J. Med. Chem., 2010, 45, 2981–2986 CrossRef CAS PubMed.
- R. F. M. Elshaarawy and C. Janiak, Eur. J. Med. Chem., 2014, 75, 31–42 CrossRef CAS PubMed.
- B. S. Creaven, D. A. Egan, D. Karcz, K. Kavanagh, M. McCann, M. Mahon, A. Noble, B. Thati and M. Walsh, J. Inorg. Biochem., 2007, 101, 1108–1119 CrossRef CAS PubMed.
- I. Georgieva, T. Mihaylov and N. Trendafilova, J. Inorg. Biochem., 2014, 135, 100–112 CrossRef CAS PubMed.
- R. B. Dixit, S. F. Vanparia, T. S. Patel, C. L. Jagani, H. V. Doshi and B. C. Dixit, Appl. Organomet. Chem., 2010, 24, 408–413 CAS.
- N. Nishat, T. Ahamad, S. M. Alshehri and S. Parveen, Eur. J. Med. Chem., 2010, 45, 1287–1294 CrossRef CAS PubMed.
- T. A. Yousef, G. M. A. El-Reash, M. Al-Jahdali and E. R. El-Rakhawy, J. Mol. Struct., 2013, 1053, 15–21 CrossRef CAS PubMed.
- M. Devereux, M. McCann, V. Leon, R. Kelly, D. O. Shea and V. McKee, Polyhedron, 2003, 22, 3187–3194 CrossRef CAS.
- N. J. S. Costa, C. R. Bombardieri, J. S. Kuribayashi, M. M. de Camargo, L. H. Andrade, E. Kagohara and B. P. Esposito, Chem. Biodiversity, 2008, 5, 2156–2159 CAS.
- L. Jin, F. Xiao, G. Cheng and Z. Ji, Inorg. Chem. Commun., 2006, 9, 758–760 CrossRef CAS PubMed.
- L. Jin, F. Xiao, G. Cheng and Z. Ji, J. Organomet. Chem., 2006, 691, 2909–2914 CrossRef CAS PubMed.
- D. P. Singh, K. Kumar and C. Sharma, Eur. J. Med. Chem., 2009, 44, 3299–3304 CrossRef CAS PubMed.
- R. V. Singh and A. Chaudhary, J. Inorg. Biochem., 2004, 98, 1712–1721 CrossRef CAS PubMed.
- I. Turel, Coord. Chem. Rev., 2002, 232, 27–47 CrossRef CAS and references cited therein.
- The Quinolones, ed. V. T. Andriole, Academic Press, 2000 Search PubMed.
- D. E. King, R. Malone and S. H. Lilley, Am. Fam. Physician, 2000, 61, 2741–2748 CAS.
- J. H. Morais Cabral, A. P. Jackson, C. V. Smith, N. Shikotra, A. Maxwell and R. C. Liddington, Nature, 1997, 388, 903–906 CrossRef CAS PubMed.
- M. E. Katsarou, E. K. Efthimiadou, G. Psomas, A. Karaliota and D. Vourloumis, J. Med. Chem., 2008, 51, 470–478 CrossRef CAS PubMed.
- I. Turel, J. Kljun, F. Perdih, E. Morozova, V. Bakulev, N. Kasyanenko, J. A. W. Byl and N. Osheroff, Inorg. Chem., 2010, 49, 10750–10752 CrossRef CAS PubMed.
- J. Kljun, A. K. Bytzek, W. Kandioller, C. Bartel, M. A. Jakupec, C. G. Hartinger, B. K. Keppler and I. Turel, Organometallics, 2011, 30, 2506–2512 CrossRef CAS PubMed.
- I. Turel, A. Golobic, A. Klavzar, B. Pihlar, P. Buglyo, E. Tolis, D. Rehder and K. Sepcic, J. Inorg. Biochem., 2003, 95, 199–207 CrossRef CAS.
- M. P. Lopez-Gresa, R. Ortiz, L. Perello, J. Latorre, M. Liu-Gonzalez, S. Garcia-Granda, M. Perez-Priede and E. Canton, J. Inorg. Biochem., 2002, 92, 65–74 CrossRef CAS.
- N. Jimenez-Garrido, L. Perello, R. Ortiz, G. Alzuet, M. Gonzalez-Alvarez, E. Canton, M. Liu-Gonzalez, S. Garcia-Granda and M. Perez-Priede, J. Inorg. Biochem., 2005, 99, 677–689 CrossRef CAS PubMed.
- P. Drevensek, I. Turel and N. Poklar Ulrih, J. Inorg. Biochem., 2003, 96, 407–415 CrossRef CAS.
- K. C. Skyrianou, F. Perdih, I. Turel, D. P. Kessissoglou and G. Psomas, J. Inorg. Biochem., 2011, 104, 63–74 CrossRef.
- G. Psomas and D. P. Kessissoglou, Dalton Trans., 2013, 42, 6252–6276 RSC.
- Meyler's Side Effects of Drugs: The International Encyclopedia of Adverse Drug Reactions and Interactions, in Sparfloxacin, ed. J. K. Aronson, Elsevier, 2006, pp. 3172–3174 Search PubMed.
- A. Tarushi, E. Polatoglou, J. Kljun, I. Turel, G. Psomas and D. P. Kessissoglou, Dalton Trans., 2011, 40, 9461–9473 RSC.
- K. C. Skyrianou, C. P. Raptopoulou, V. Psycharis, D. P. Kessissoglou and G. Psomas, Polyhedron, 2009, 28, 3265–3271 CrossRef CAS PubMed.
- K. C. Skyrianou, E. K. Efthimiadou, V. Psycharis, A. Terzis, D. P. Kessissoglou and G. Psomas, J. Inorg. Biochem., 2009, 103, 1617–1625 CrossRef CAS PubMed.
- E. K. Efthimiadou, Y. Sanakis, C. P. Raptopoulou, A. Karaliota, N. Katsaros and G. Psomas, Bioorg. Med. Chem. Lett., 2006, 16, 3864–3867 CrossRef CAS PubMed.
- D. Shingnapurkar, R. Butcher, Z. Afrasiabi, E. Sinn, F. Ahmed, F. Sarkar and S. Padhye, Inorg. Chem. Commun., 2007, 10, 459–462 CrossRef CAS PubMed.
- C. Tan, J. Liu, H. Li, W. Zheng, S. Shi, L. Chen and L. Ji, J. Inorg. Biochem., 2008, 102, 347–358 CrossRef CAS PubMed.
- J. Marmur, J. Mol. Biol., 1961, 3, 208–211 CrossRef CAS.
- M. F. Reichmann, S. A. Rice, C. A. Thomas and P. Doty, J. Am. Chem. Soc., 1954, 76, 3047–3053 CrossRef CAS.
- Agilent, CrysAlis PRO, Agilent Technologies, Yarnton, Oxfordshire, England, 2013 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- J. M. Andrews, J. Antimicrob. Chemother., 2001, 48(suppl. S1), 5–16 CrossRef CAS PubMed.
- A. Wolfe, G. Shimer and T. Meehan, Biochemistry, 1987, 26, 6392–6396 CrossRef CAS.
- Y. Wang, H. Zhang, G. Zhang, W. Tao and S. Tang, J. Lumin., 2007, 126, 211–218 CrossRef CAS PubMed.
- L. Stella, A. L. Capodilupo and M. Bietti, Chem. Commun., 2008, 4744–4746 RSC.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Part B: Applications in Coordination, Organometallic, and Bioinorganic Chemistry, Wiley, New Jersey, 6th edn, 2009 Search PubMed.
- M. Zampakou, N. Rizeq, V. Tangoulis, A. N. Papadopoulos, F. Perdih, I. Turel and G. Psomas, Inorg. Chem., 2014, 53, 2040–2052 CrossRef CAS PubMed.
- B. Chiswell, E. D. McKenzie and L. F. Lindoy, in Comprehensive Coordination Chemistry, ed. G. Wilkinson, Pergamon Press, Oxford, 1987, vol. 4, pp. 1–122 Search PubMed.
- B. A. Frenz and J. A. Ibers, Inorg. Chem., 1972, 11, 1109–1116 CrossRef CAS.
- A. Grirrane, A. Pastor, A. Ienco, C. Mealli and A. Galindo, J. Chem. Soc., Dalton Trans., 2002, 3771–3777 RSC.
- C. Protogeraki, E. G. Andreadou, F. Perdih, I. Turel, A. A. Pantazaki and G. Psomas, Eur. J. Med. Chem., 2014, 86, 189–201 CrossRef CAS PubMed.
- A. Tarushi, G. Psomas, C. P. Raptopoulou, V. Psycharis and D. P. Kessissoglou, Polyhedron, 2009, 28, 3272–3278 CrossRef CAS PubMed.
- V. Uivarosi, Molecules, 2013, 18, 11153–11197 CrossRef CAS PubMed.
- A. D. Russell, in Disinfection, Sterilization and Preservation, ed. S. S. Block, Lippincott Williams & Wilkins, Philadelphia, 5th edn, 2001, pp. 31–56 Search PubMed.
- N. Ahmed, M. Dawson, C. Smith and E. Wood, Biology of Disease, Taylor & Francis, 2007, ch. 3, pp. 41–71 Search PubMed.
- J. Kljun, I. Bratsos, E. Alessio, G. Psomas, U. Repnik, M. Butinar, B. Turk and I. Turel, Inorg. Chem., 2013, 52, 9039–9052 CrossRef CAS PubMed.
- M. M. Aleksic and V. Kapetanovic, Acta Chim. Slov., 2014, 61, 555–573 CAS.
- G. Pratviel, J. Bernadou and B. Meunier, Adv. Inorg. Chem., 1998, 45, 251–262 CrossRef CAS.
- A. Dimitrakopoulou, C. Dendrinou-Samara, A. A. Pantazaki, M. Alexiou, E. Nordlander and D. P. Kessissoglou, J. Inorg. Biochem., 2008, 102, 618–628 CrossRef CAS PubMed.
- W. D. Wilson, L. Ratmeyer, M. Zhao, L. Strekowski and D. Boykin, Biochemistry, 1993, 32, 4098–4104 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- W. D. Wilson, L. Ratmeyer, M. Zhao, L. Strekowski and D. Boykin, Biochemistry, 1993, 32, 4098–4104 CrossRef CAS.
- O. H. Laitinen, V. P. Hytonen, H. R. Nordlund and M. S. Kulomaa, Cell. Mol. Life Sci., 2006, 63, 2992–3017 CrossRef CAS PubMed.
- V. Rajendiran, R. Karthik, M. Palaniandavar, H. Stoeckli-Evans, V. S. Periasamy, M. A. Akbarsha, B. S. Srinag and H. Krishnamurthy, Inorg. Chem., 2007, 46, 8208–8221 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1026288. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra11682h |
| This journal is © The Royal Society of Chemistry 2015 |