
Defect driven ferromagnetism in SnO2: a combined study using density functional theory and positron annihilation spectroscopy
Abstract
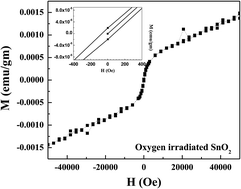
Room temperature ferromagnetic ordering has been observed in a high purity polycrystalline SnO2 sample due to irradiation of 96 MeV oxygen ions. Ab initio density functional theory calculation indicates that tin vacancies are mainly responsible for inducing the magnetic moment in SnO2 whereas oxygen vacancies in SnO2 do not contribute any magnetic moment. Positron annihilation spectroscopy has been employed to characterize the chemical identity of irradiation generated defects in SnO2. Results indicate the dominant presence of Sn vacancies in O ion irradiated SnO2. The irradiated sample turns out to be ferromagnetic at room temperature.
 Please wait while we load your content...
Please wait while we load your content...

