
Flexible and ultralong-life cuprous oxide microsphere-nanosheets with superior pseudocapacitive properties
Chaoqun Donga,
Qingguo Baia,
Guanhua Chenga,
Bingge Zhaob,
Hao Wanga,
Yulai Gaob and
Zhonghua Zhang*a
aKey Laboratory for Liquid-Solid Structural Evolution and Processing of Materials (Ministry of Education), School of Materials Science and Engineering, Shandong University, Jingshi Road 17923, Jinan 250061, P.R. China. E-mail: zh_zhang@sdu.edu.cn; Fax: +86-531-88396978; Tel: +86-531-88396978
bLaboratory for Microstructures, Shanghai University, 99 Shangda Road, Shanghai 200436, P. R. China
First published on 15th December 2014
Abstract
Nanostructured transition metal oxides have been investigated extensively for supercapacitor electrodes due to their high theoretical specific capacitance, low-cost, environment benignity and abundance. However, pristine transition metal oxides suffer from difficulty of synthesis, poor electronic conductivity and mechanical flexibility. In this work, we report a facile, low-cost and high-throughput synthesis of hierarchical structure which consists of cuprous oxide (Cu2O) microsphere-nanosheets on the surface of flexible Cu foil (namely Cu2O@Cu) via a two-step electrochemical method (anodization and electro-oxidation). The influence of the anodization parameters on surface roughness of Cu foil has been investigated, and the optimum anodization procedure was determined to be 50 V for 4 cycles. This Cu2O@Cu electrode exhibits excellent capacitance properties, such as up to 390.9 mF cm−2 at 2 mA cm−2 in areal capacitance, and high flexibility, as observed by cyclic voltammetry measurement under various deformation (bending and folding) situations. Furthermore, the Cu2O@Cu electrode presents superior long-term cycling stability over 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles, with the capacitance retention of over 80%. The present binder-free Cu2O@Cu microsphere-nanosheets electrode is highly promising for future applications in flexible supercapacitors.
000 cycles, with the capacitance retention of over 80%. The present binder-free Cu2O@Cu microsphere-nanosheets electrode is highly promising for future applications in flexible supercapacitors.
Introduction
The increasing demand for next-generation portable electronics with roll-up and wearable characteristics has inspired intensive efforts to explore high-performance energy storage devices that are flexible, lightweight, and environmentally friendly.1–3 Among these, supercapacitors have been widely investigated as one of the most important and advanced candidates due to their high power density, ultra-fast charge/discharge rate, superlong cycling life, and then enable highly efficient, safe, green use of energy.4–7 Among various supercapacitor electrode materials, nanostructured transition metal oxides, such as RuO2,8 MnO2,9 Co3O4,10 TiO2,11 NiO,12 Fe3O4,13 V2O5,14 SnO215 which possess multiple oxidation states that enable rich redox reactions for pseudocapacitance generation, can provide higher specific capacitance and energy density than that delivered by carbon-based active materials, and better cycling stability than conductive polymer materials.16 However, the undesirably poor electronic conductivity of metal oxides significantly impedes them from wide use in supercapacitor devices.17 One of major strategies to enhance charge transport in electrodes is to design composite materials by combining metal oxides with a conductive substance.17 Carbon in various forms18–20 and conductive polymers21–23 have been explored to serve as conductive pathways of metal oxides. Although these composites could improve electronic conductivity, the assembled electrodes still have to face the deficiency of high contact resistance between the metal oxides and the current collector, greatly hindering their practical applications.24Besides these issues, conventional supercapacitors with current collectors, insulating binder and conductive additives are still too thick, bulky and rigid to meet the practical requirement for flexible energy storage devices.25,26 A considerable part of these electrodes are susceptible to structure fracture and the active materials are apt to peel off under the situation of bending or during charge/discharge process, giving rise to abundant waste, inconvenience, as well as safety problems.27 Thus, there is an urgent need to develop flexible or even foldable supercapacitors which can accommodate large levels of deformation without sacrificing electrochemical performance.28 Recently, flexible supercapacitor electrodes were mainly fabricated by integrating active capacitive materials including metal oxides, conductive polymers, and carbon materials, into various flexible substrates, such as cellulose paper, textiles, plastics and carbon fiber paper, exhibiting remarkable flexibility.29–33 However, cellulose paper, textiles and plastics are typical insulators and much effort should be dedicated to enhance the conductivity of them.34 Moreover, cellulose paper is easy to be torn up, resulting in the whole scale breakdown of the supercapacitor. As for carbon fiber paper, its specific capacitance is much lower when comparing to metal oxides.
In the past several years, cuprous oxide (Cu2O) has been synthesized through various routes and extensively studied for applications in Li-ion batteries.35,36 Xiang et al.37 fabricated core–shell Cu2O/Cu composites towards Li-ion batteries application by a special reduction process of CuSO4. Park et al.38 synthesized uniform Cu2O nanocubes in a gram scale by a one-pot polyol process. Besides, many other groups have synthesized Cu2O for Li-ion batteries by using γ-irradiation,39 thermal decomposition,40 electrochemical oxidation,41 and chemical bath deposition42 etc., showing remarkable charge storage capacity. These existing results well suggest promising charge-storage performance of Cu2O for pseudocapacitors.
Based on the above considerations, in this study, we report a facile electrochemical route to prepare cuprous oxide (Cu2O) microsphere-nanosheets which in situ grow on the surface of Cu foil substrate, and exhibit superior pseudocapacitive performance. The aims of designing such an electrode are listed as follows: (i) Cu foil is quite abundant, flexible, thin, cheap and environmentally friendly; (ii) Cu2O in situ grows on the surface of Cu foil without any binders and conductive additives, which can effectively ensure fine interface contact between the active material and substrate, as well as excellent conductivity of the whole electrode; (iii) Cu foil can not only serve as the support for the active material but serve as the current collector, which greatly reduces the size of supercapacitors; (iv) compared with various reported flexible composite electrodes, the fabricating method of the Cu2O@Cu electrode is facile, safe, environmentally friendly and high-throughput. Here, the flexible Cu2O@Cu electrode exhibits high areal capacitance and superior long-term cycling stability, suggesting promising future in practical applications for supercapacitors.
Experimental section
The Cu2O@Cu microsphere-nanosheets electrode was fabricated through a two-step electrochemical process, namely the anodization of Cu foil to prepare nanostructured Cu followed by electro-oxidation to form Cu2O microsphere-nanosheets. The anodization procedure was operated in a two-electrode electrochemical cell with a commercial Cu foil and a graphite plate as the anode and cathode electrode, respectively. Both the two electrodes were sealed to ensure that the geometric working area was 1.0 cm × 1.0 cm. Anodization was carried out in an aqueous solution containing 0.4 M H2C2O4 and a little amount of HF at a constant potential (10, 20, 30, 40 and 50 V) for 3 min, and then −10 V for 30 s at room temperature. This was regarded as 1 cycle, while the anodization needed to cycle several times. A potentiostat (CHI660E, Shanghai, Chenhua) with a three-electrode configuration was used to perform electro-oxidation. The as-prepared nanostructured Cu foil was used as the working electrode, a Pt foil (1 cm × 1 cm) acted as the counter electrode and an Ag/AgCl electrode served as the reference electrode. The Cu2O microsphere-nanosheets were fabricated by employing cyclic voltammetry (CV) in the potential range from −0.4 to 0.4 V vs. Ag/AgCl at the scan rate of 5 mV s−1 for 1 segment in a 1 M KOH aqueous solution. The obtained electrodes were designated as Cu2O@Cu for simplicity.The phase formation of the as-synthesized samples was characterized using X-ray diffraction (XRD, Rigaku D/max-rB) with Cu Kα radiation. The size and morphology of the samples were characterized using a scanning electron microscope (SEM, LEO 1530 VP). The resistivity of Cu foil and the Cu2O@Cu electrode was measured by a constant current four-electrode method, and was recorded every 2 s by a computer connected to the measurement system. Nitrogen adsorption/desorption isotherms were measured at 77 K with a V-Sorb 2800P Surface Area and Pore Size Analyzer. The specific surface area was calculated using the Brunauer–Emmett–Teller (BET) method.43–45 The pore size distribution was obtained by the Barrett–Joyner–Halenda (BJH) method.10,46,47
The mass of Cu2O was calculated by means of the following steps. Firstly, the mass of the completely clean and dried Cu2O@Cu electrode was weighed, and named m1. Secondly, this electrode was immersed into a N2-saturated 0.1 M HCl solution about 10 min until no obvious color change occurred. During this step, the Cu2O layer was removed. The mass of the Cu foil, named m2, was weighed again after careful washing with highly pure de-ionized water and complete drying. Finally, the mass of Cu2O attached on Cu foil was calculated through (m1 − m2).
To evaluate the pseudocapacitive performance of the Cu2O@Cu electrode, all electrochemical measurements were carried out using the potentiostat (CHI660E, Shanghai, Chenhua) in a three-electrode electrochemical cell. The Cu2O@Cu electrode was used as the working electrode, with the Pt foil as the counter electrode and the Ag/AgCl electrode as the reference electrode. All electrochemical tests (CV and galvanostatic charge/discharge) were performed in a 1 M KOH aqueous solution at room temperature.
Results and discussion
The Cu2O@Cu electrode was developed through the anodization of Cu foil followed by electro-oxidation to form Cu2O microsphere-nanosheets. Compared to common methods, including template-directed,9,48,49 hydrothermal,50,51 or solvothermal,52,53 as well as dealloying preparation54–56 for nanomaterials, the proposed electrochemistry strategy does not involve specific templates, high temperatures, reducing agents, and separation/purification procedures. To study the effect of anodizing voltage on the electrochemical performance and further explore the optimum voltage for anodization, the Cu foil samples were anodized at 10 V, 20 V, 30 V, 40 V and 50 V for 1 cycle respectively, and then were electro-oxidized to Cu2O. The pseudocapacitive performance of these Cu2O@Cu electrodes were measured by calculating the loop area recorded using cyclic voltammetry in the potential range from 0.3 to 0.7 V (vs. Ag/AgCl). Here for comparison, the surface increasing factor (SIF) for Cu2O@Cu electrode gained only by electro-oxidation (without anodization) was defined to be 1. Fig. 1a shows the SIFs for the Cu2O@Cu electrodes subjected to anodization under different voltages for one cycle and the same electro-oxidation process. As we can see, the value of SIF increases with increasing anodizing voltage. It is worth noting that the increasing degree of SIF is not significant as the anodizing voltage varies from 10 V to 40 V, while the value of SIF increases sharply as the voltage further reaches up to 50 V. The SIF at 50 V is 43.2, indicating that the surface of Cu foil was roughened to a large extent after anodization at 50 V and the loading of Cu2O would increase dramatically after the subsequent electro-oxidation. Similarly, the Cu2O@Cu electrodes fabricated through anodization at 50 V for various cycles followed by the same electro-oxidation were used to explore the optimal preparation process. Fig. 1b displays the relationship between the SIF values and the number of anodizing cycles. The SIF is enhanced obviously with the increasing number of anodizing cycles at first (less than 4 cycles), while too many cycles will weaken the surface roughness of Cu foil. It is mainly due to the roughed Cu surface layer will peel off when cycling too many times. As expected, the SIF at 50 V for 4 cycles reaches up to 304. Finally, the optimum anodization procedure was determined to be 50 V for 4 cycles.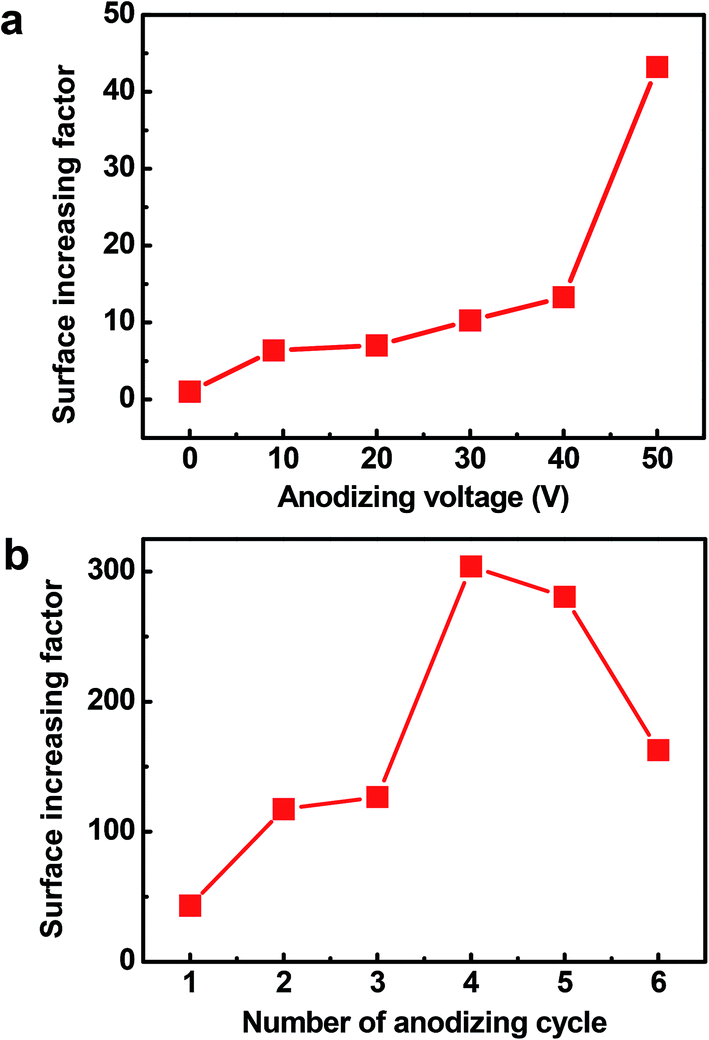 | ||
| Fig. 1 (a) Surface increasing factors (SIFs) for the Cu2O@Cu electrodes subjected to anodization for 1 cycle under different anodizing voltages and the same electro-oxidation. (b) SIFs for the Cu2O@Cu electrodes subjected to anodization under 50 V for various cycles and the same electro-oxidation. | ||
Fig. 2 depicts the SEM images of the Cu foil electrode after anodization at 50 V for 4 cycles. It can be seen that irregular-shaped Cu particles disperse well on the surface of the Cu foil, which gives rise to the rough island-like surface and remarkable enhanced specific surface area. The size of the Cu particles is submicron. Fig. 3 illustrates the low- and high-magnification SEM images of the Cu2O@Cu electrode obtained through the anodization (50 V/4 cycles) and subsequent electro-oxidation. The surface of the electrode (Fig. 3a) is ravine-like and is further roughened remarkably compared to that of the anodized Cu foil. Careful observation (Fig. 3b and c) can find that the surface is covered by microspheres consisting of nanosheets stretching along different directions. The nanosheets with about 20 nm in thickness are interconnected with each other, and one nanosheet is highlighted by an arrow in the inset of Fig. 3c. There exist micron-sized channels among the microspheres as well as nano-sized channels among the nanosheets, which could allow easy diffusion of the electrolyte onto the nanosheets surface. Fig. 4 shows the corresponding X-ray diffraction (XRD) pattern of the Cu2O@Cu electrode. The diffraction peaks at 2θ of 36.4° and 42.4° are attributed to the (111) and (200) reflections of Cu2O (JCPDS no. 05-0667) and the three characteristic peaks at 2θ of 43.3°, 50.5° and 74.2° correspond to the (111), (200) and (220) planes of Cu (JCPDS no. 04-0836).36,37,39 The weak (200) peak of Cu2O is highlighted as the inset in Fig. 4. The present results well confirm the formation of the Cu2O microsphere-nanosheets on the Cu foil substrate.
 | ||
| Fig. 2 Low- and high-magnification SEM images of the Cu foil electrode after anodization at 50 V for 4 cycles. | ||
 | ||
| Fig. 3 Low- and high-magnification SEM images of the Cu2O@Cu electrode. | ||
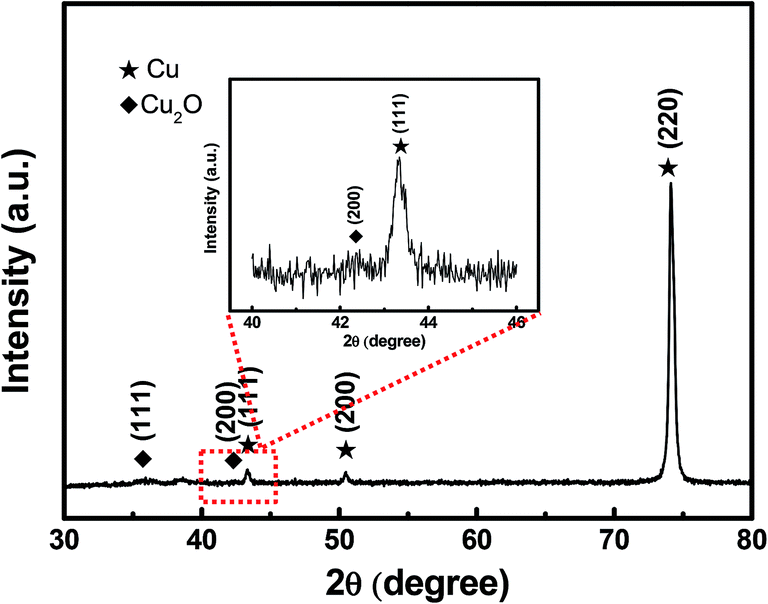 | ||
| Fig. 4 XRD pattern of the Cu2O@Cu nanosheets electrode. | ||
The specific surface area and porous structure of the as-prepared Cu2O@Cu sample was investigated by N2 adsorption/desorption measurements. The N2 adsorption/desorption isotherm of the sample (Fig. 5a) can be classified as a type IV isotherm with a H3-type hysteresis loop, indicating the existence of mesopores possibly formed by the loose stacking of Cu2O nanosheets.57,58 On the basis of the adsorption/desorption isotherm, the BET surface area of the Cu2O@Cu electrode was calculated to be 0.5997 m2 g−1 (considering the whole mass of Cu2O active material and Cu foil substrate), which is 250 times that of raw Cu foil (only 0.0024 m2 g−1). Fig. 5b presents the pore size distribution calculated from the N2 adsorption/desorption isotherm using the BJH method. The average pore diameter of the Cu2O@Cu electrode is in the mesoporous region, with multimodal pore size distributions. The pore size distribution maximums of the sample mainly center in the range of 2–10 nm. Combined with the above results, it suggests that such type of hierarchical surface morphologies with well-developed pore structures possess the advantages of both microsized structures and nanostructures, which can not only offer a large electrochemical surface area, but also provide good electrolyte access.59,60 The electrolyte could come into contact with more active surfaces through abundant pores and channels, which can enable excellent electrochemical behaviors of the Cu2O@Cu microsphere-nanosheets electrode.
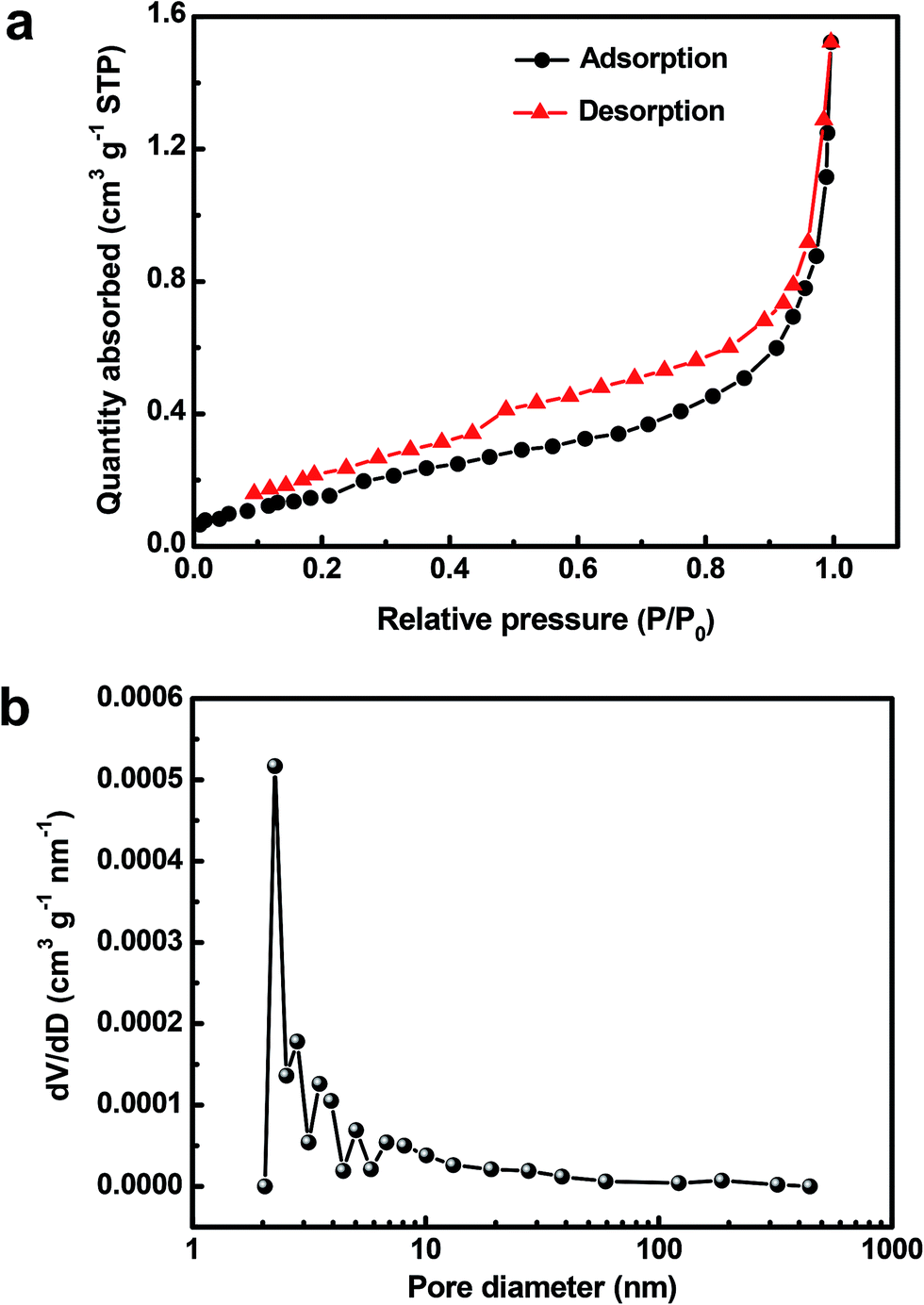 | ||
| Fig. 5 (a) N2 adsorption–desorption isotherm and (b) BJH adsorption pore size distribution of the Cu2O@Cu nanosheets electrode. | ||
To explore the electrochemical performance of the binder-free Cu2O@Cu microsphere-nanosheets electrode for supercapacitors, cyclic voltammetry (CV), galvanostatic charge/discharge and cyclic stability measurements were performed in a three-electrode cell. Fig. 6a shows the representative CV curves of the Cu2O@Cu microsphere-nanosheets electrode in the voltage window of 0.3 to 0.7 V (vs. Ag/AgCl) at various scan rates. All the curves present strong redox peaks and quite different from the ideal rectangular shape for electric double layer capacitance, indicating that the capacitance of the electrode primarily derives from faradic redox reactions. The anodic current flow is associated with oxidation of Cu+ to Cu2+ during the positive-going scan. Similarly, the cathodic peak is related to its reverse process during the negative-going scan. The cathodic peak current increases linearly with the increment of the scan rate (inset of Fig. 6a), suggesting that the rates of electronic and ionic transportation are rapid enough with respect to the scan rates.51 It should be noted that the cathodic peak position shifts in the more negative direction with the increase of the scan rate from 1 to 100 mV s−1. But the shape of the series CV curves has no significant change, indicative of excellent electronic conductivity of the Cu2O@Cu electrode. The conductivity of the Cu foil and Cu2O@Cu electrode was measured by a constant current four-electrode method. The calculated conductivity of the Cu foil and Cu2O@Cu electrode are 0.556 × 106 S cm−1 and 0.513 × 106 S cm−1 respectively, confirming the high conductivity of the Cu2O@Cu electrode. The areal capacitance could be calculated from CV curves by the formula,61
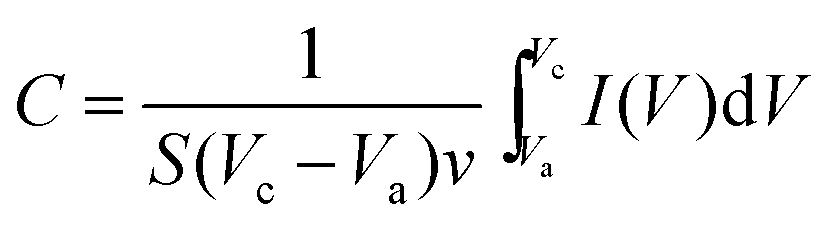 | (1) |
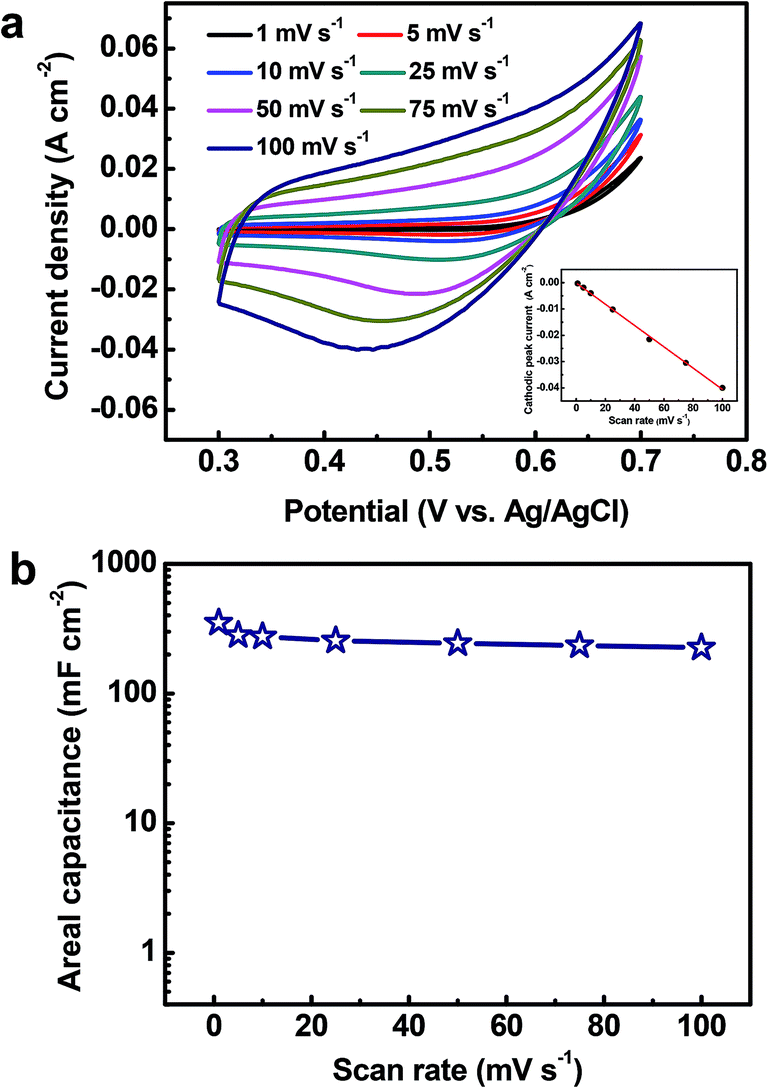 | ||
| Fig. 6 (a) CV curves of the Cu2O@Cu nanosheets electrode at various scan rates (1–100 mV s−1) recorded in the 1 M KOH aqueous solution. The inset presents plot of cathodic peak currents versus the scan rate. (b) Specific capacitance of the electrode as a function of scan rate based on the CVs. | ||
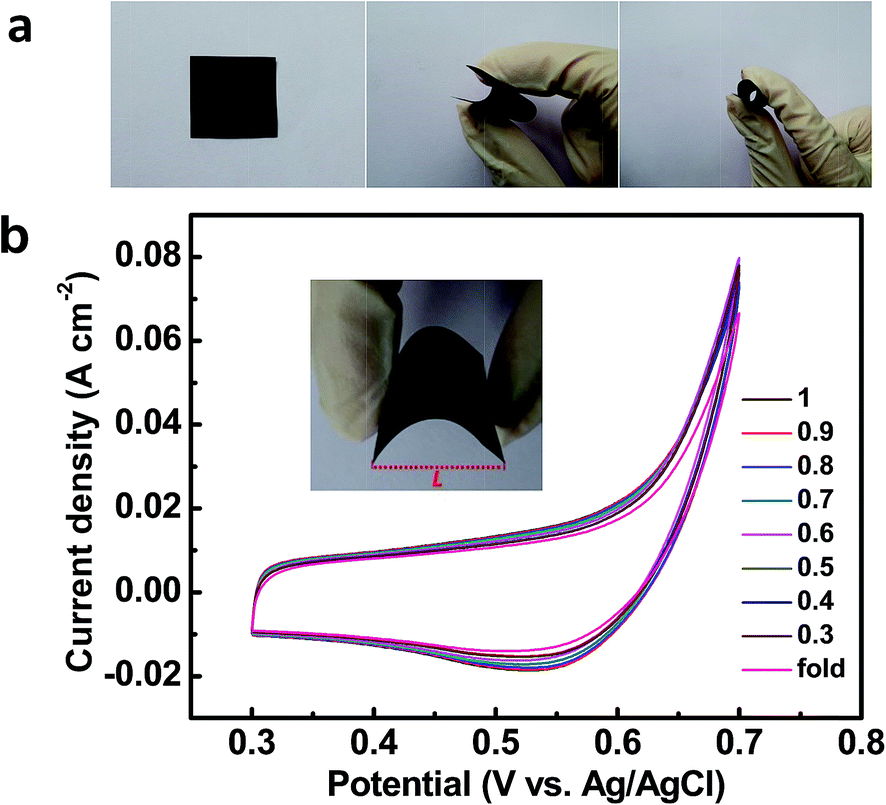 | ||
| Fig. 7 (a) Photos of the fabricated Cu2O@Cu nanosheets electrode in the flat (left), bending (middle) and fold (right) states. (b) CV curves of the Cu2O@Cu nanosheets electrode in various bending states (different L/L0 values and fold states), where L0 is the length of the electrode in the flat state, and L is the distance between two ends of the electrode in different bending states (inset). Scan rate: 50 mV s−1. | ||
Fig. 8a shows the galvanostatic charge/discharge profiles of the Cu2O@Cu microsphere-nanosheets electrode. An increase in the IR drop with an increase in the discharge current density can be clearly observed. The nonlinear feature of charge/discharge behavior further demonstrates the pseudocapacitive nature of the Cu2O@Cu microsphere-nanosheets electrode. The areal capacitance of the electrode can be calculated from the galvanostatic charge/discharge curves by the following equation:4
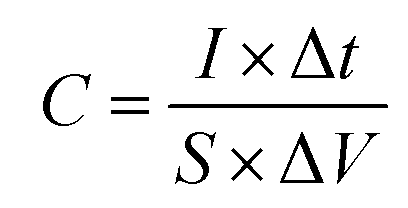 | (2) |
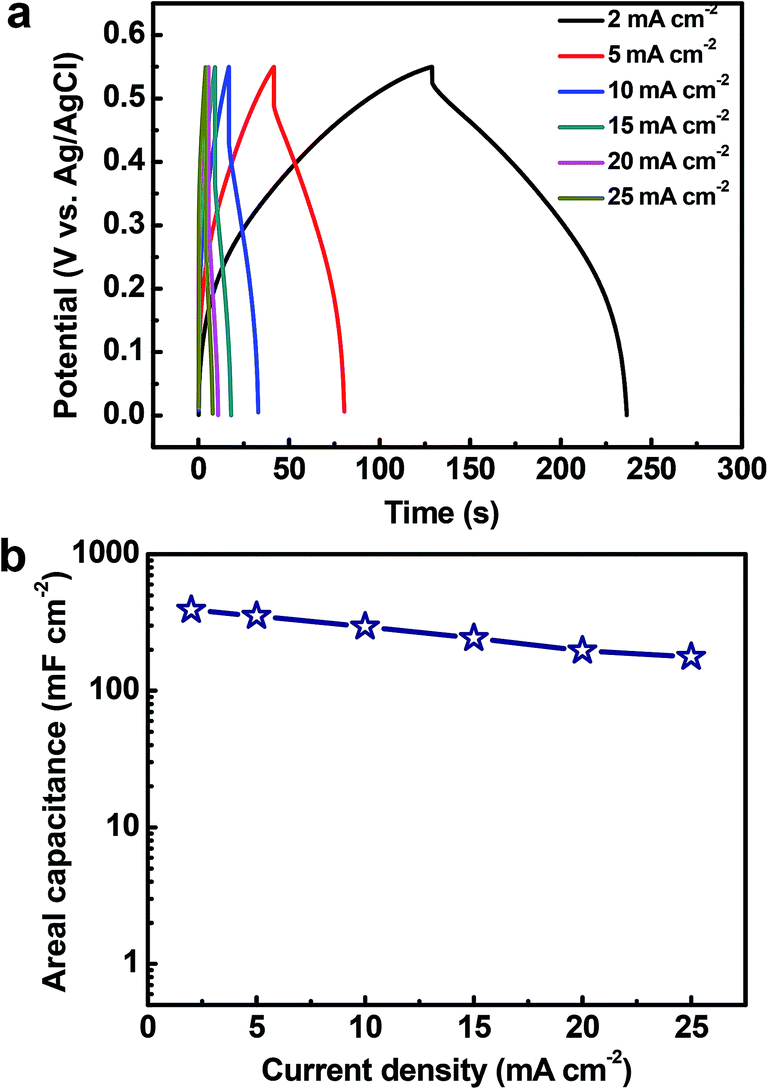 | ||
| Fig. 8 (a) Galvanostatic charge/discharge profiles of the Cu2O@Cu nanosheets electrode at different current densities. (b) The specific capacitance of the Cu2O@Cu nanosheets electrode versus the current density, based on the charge/discharge curves. | ||
The cycling stability of electrode materials is a critical requirement for practical applications. The cycling stability of the Cu2O@Cu electrode was investigated in the range of 0.3–0.7 V (vs. Ag/AgCl) in the 1 M KOH aqueous solution by repeated CV processes at the scan rate of 100 mV s−1 (Fig. 9). The areal capacitance still remains 95.8% and 80.4% of the initial value each after 50000 and 100000 cycles, respectively. To the best of our knowledge, such superior cycling stability has not been reported in the literature for metal oxides till now. After 100000 cycles, the microstructure of the electrode was observed using SEM (inset of Fig. 9). The microsphere-nanosheets morphology of the electrode still remained, but the nanosheets became thicker after 100000 cycles, which could well explain the 19.6% loss of the specific capacitance.
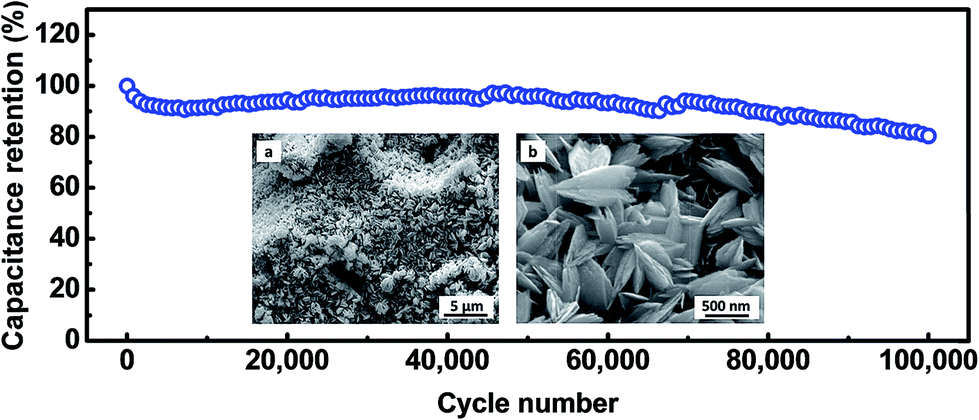 | ||
| Fig. 9 Cycling performance of the Cu2O@Cu nanosheets electrode at the scan rate of 100 mV s−1. The insets present low- and high-magnification SEM images of the Cu2O electrode after 100000 cycles. | ||
Conclusions
In summary, a Cu2O@Cu microsphere-nanosheets electrode has been developed through a facile two-step electrochemical method. The Cu2O microsphere-nanosheets in situ grow on the surface of Cu foil without any binders and conductive additives, which not only keeps fine interface contact between the active material and substrate, but also ensures excellent conductivity of the electrode. The as-prepared Cu2O@Cu electrode exhibits remarkable mechanical flexibility and high specific capacitance of 390.9 mF cm−2 at 2 mA cm−2. Moreover, the excellent cycling stability was confirmed to be about 80.4% retention of the initial capacitance after 100000 cycles. All these points suggest that the binder-free Cu2O@Cu microsphere-nanosheets electrode is promising for flexible high-performance supercapacitors.
Acknowledgements
The authors gratefully acknowledge financial support by National Natural Science Foundation of China (51371106), Specialized Research Found for the Doctoral Program of Higher Education of China (20120131110017), and Young Tip-top Talent Support Project (the Organization Department of the Central Committee of the CPC). We also acknowledge the experimental assistance from Ruhr University Bochum, Germany. Y. L. Gao acknowledges the support by Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning (no. TP2014042).References
- Y. Qin, X. Wang and Z. L. Wang, Nature, 2008, 451, 809–813 CrossRef CAS PubMed.
- K. Wang, Q. Meng, Y. Zhang, Z. Wei and M. Miao, Adv. Mater., 2013, 25, 1494–1498 CrossRef CAS PubMed.
- L. Yuan, B. Yao, B. Hu, K. Huo, W. Chen and J. Zhou, Energy Environ. Sci., 2013, 6, 470–476 CAS.
- P. Yang, Y. Ding, Z. Lin, Z. Chen, Y. Li, P. Qiang, M. Ebrahimi, W. Mai, C. P. Wong and Z. L. Wang, Nano Lett., 2014, 14, 731–736 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed.
- X. Wang, C. Yan, A. Sumboja and P. S. Lee, Nano Energy, 2014, 3, 119–126 CrossRef CAS PubMed.
- Y.-Z. Su, K. Xiao, N. Li, Z.-Q. Liu and S.-Z. Qiao, J. Mater. Chem. A, 2014, 2, 13845–13853 CAS.
- R.-R. Bi, X.-L. Wu, F.-F. Cao, L.-Y. Jiang, Y.-G. Guo and L.-J. Wan, J. Phys. Chem. C, 2010, 114, 2448–2451 CAS.
- Z. Yu, B. Duong, D. Abbitt and J. Thomas, Adv. Mater., 2013, 25, 3302–3306 CrossRef CAS PubMed.
- R. B. Rakhi, W. Chen, M. N. Hedhili, D. Cha and H. N. Alshareef, ACS Appl. Mater. Interfaces, 2014, 6, 4196–4206 CAS.
- X. Lu, G. Wang, T. Zhai, M. Yu, J. Gan, Y. Tong and Y. Li, Nano Lett., 2012, 12, 1690–1696 CrossRef CAS PubMed.
- G. Zhang, L. Yu, H. E. Hoster and X. W. Lou, Nanoscale, 2013, 5, 877–881 RSC.
- J. Mu, B. Chen, Z. Guo, M. Zhang, Z. Zhang, P. Zhang, C. Shao and Y. Liu, Nanoscale, 2011, 3, 5034–5040 RSC.
- B. Saravanakumar, K. K. Purushothaman and G. Muralidharan, ACS Appl. Mater. Interfaces, 2012, 4, 4484–4490 CAS.
- W. Wang, Q. Hao, W. Lei, X. Xia and X. Wang, RSC Adv., 2012, 2, 10268–10274 RSC.
- F. Wang, S. Xiao, Y. Hou, C. Hu, L. Liu and Y. Wu, RSC Adv., 2013, 3, 13059–13084 RSC.
- M. Zhi, C. Xiang, J. Li, M. Li and N. Wu, Nanoscale, 2013, 5, 72–88 RSC.
- C. Wu, S. Deng, H. Wang, Y. Sun, J. Liu and H. Yan, ACS Appl. Mater. Interfaces, 2014, 6, 1106–1112 CAS.
- Q. Li, X.-F. Lu, H. Xu, Y.-X. Tong and G.-R. Li, ACS Appl. Mater. Interfaces, 2014, 6, 2726–2733 CAS.
- P. Li, Y. Yang, E. Shi, Q. Shen, Y. Shang, S. Wu, J. Wei, K. Wang, H. Zhu, Q. Yuan, A. Cao and D. Wu, ACS Appl. Mater. Interfaces, 2014, 6, 5228–5234 CAS.
- C. Zhou, Y. Zhang, Y. Li and J. Liu, Nano Lett., 2013, 13, 2078–2085 CrossRef CAS PubMed.
- Q. Qu, Y. Zhu, X. Gao and Y. Wu, Adv. Energy Mater., 2012, 2, 950–955 CrossRef CAS.
- Y. Hou, L. Chen, P. Liu, J. Kang, T. Fujita and M. Chen, J. Mater. Chem. A, 2014, 2, 10910–10916 CAS.
- L. Yu, Y. Jin, L. Li, J. Ma, G. Wang, B. Geng and X. Zhang, CrystEngComm, 2013, 15, 7657–7662 RSC.
- J. P. Cheng, X. Chen, J.-S. Wu, F. Liu, X. B. Zhang and V. P. Dravid, CrystEngComm, 2012, 14, 6702–6709 RSC.
- L. Fan, L. Tang, H. Gong, Z. Yao and R. Guo, J. Mater. Chem., 2012, 22, 16376–16381 RSC.
- H. Wang, B. Zhu, W. Jiang, Y. Yang, W. R. Leow, H. Wang and X. Chen, Adv. Mater., 2014, 26, 3638–3643 CrossRef CAS PubMed.
- X. Dong, Z. Guo, Y. Song, M. Hou, J. Wang, Y. Wang and Y. Xia, Adv. Funct. Mater., 2014, 24, 3405–3412 CrossRef CAS.
- Y.-C. Chen, Y.-K. Hsu, Y.-G. Lin, Y.-K. Lin, Y.-Y. Horng, L.-C. Chen and K.-H. Chen, Electrochim. Acta, 2011, 56, 7124–7130 CrossRef CAS PubMed.
- S.-L. Chou, J.-Z. Wang, S.-Y. Chew, H.-K. Liu and S.-X. Dou, Electrochem. Commun., 2008, 10, 1724–1727 CrossRef CAS PubMed.
- Y. Zhao, Y. Meng and P. Jiang, J. Power Sources, 2014, 259, 219–226 CrossRef CAS PubMed.
- I. Shakir, Z. Ali, J. Bae, J. Park and D. J. Kang, Nanoscale, 2014, 6, 4125–4130 RSC.
- C. Ma, Y. Li, J. Shi, Y. Song and L. Liu, Chem. Eng. J., 2014, 249, 216–225 CrossRef CAS PubMed.
- J.-X. Feng, Q. Li, X.-F. Lu, Y.-X. Tong and G.-R. Li, J. Mater. Chem. A, 2014, 2, 2985–2992 CAS.
- K. Chen and D. Xue, Funct. Mater. Lett., 2014, 7, 1430001 CrossRef (9 pages).
- Q. Pan and M. Wang, J. Electrochem. Soc., 2008, 155, A452–A457 CrossRef CAS PubMed.
- J. Y. Xiang, J. P. Tu, Y. F. Yuan, X. H. Huang, Y. Zhou and L. Zhang, Electrochem. Commun., 2009, 11, 262–265 CrossRef CAS PubMed.
- J. C. Park, J. Kim, H. Kwon and H. Song, Adv. Mater., 2009, 21, 803–807 CrossRef CAS.
- W. Liu, G. Chen, G. He and W. Zhang, J. Nanopart. Res., 2011, 13, 2705–2713 CrossRef CAS.
- L. Hu, Y. Huang, F. Zhang and Q. Chen, Nanoscale, 2013, 5, 4186–4190 RSC.
- J. Y. Xiang, J. P. Tu, X. H. Huang and Y. Z. Yang, J. Solid State Electrochem., 2008, 12, 941–945 CrossRef CAS PubMed.
- Q. Pan, M. Wang and Z. Wang, Electrochem. Solid-State Lett., 2009, 12, A50–A53 CrossRef CAS PubMed.
- A. Seri-Levy and D. Avnir, Langmuir, 1993, 9, 2523–2529 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- K. Sing, Colloids Surf., A, 2001, 187–188, 3–9 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- J. Choma, M. Jaroniec, W. Burakiewicz-Mortka and M. Kloske, Appl. Surf. Sci., 2002, 196, 216–223 CrossRef CAS.
- C. Wang, G. Zhou, D. Xu, B. Sun, Y. Zhang and F. Chen, RSC Adv., 2014, 4, 37270–37273 RSC.
- N. Du, H. Zhang, B. Chen, X. Ma and D. Yang, Chem. Commun., 2008, 3028–3030 RSC.
- X. Feng, N. Chen, Y. Zhang, Z. Yan, X. Liu, Y. Ma, Q. Shen, L. Wang and W. Huang, J. Mater. Chem. A, 2014, 2, 9178–9184 CAS.
- X. Yu, B. Lu and Z. Xu, Adv. Mater., 2014, 26, 1044–1051 CrossRef CAS PubMed.
- L. Hu, B. Qiu, Y. Xia, Z. Qin, L. Qin, X. Zhou and Z. Liu, J. Power Sources, 2014, 248, 246–252 CrossRef CAS PubMed.
- H. Akram, C. Mateos-Pedrero, E. Gallegos-Suárez, A. Guerrero-Ruíz, T. Chafik and I. Rodríguez-Ramos, Appl. Surf. Sci., 2014, 307, 319–326 CrossRef CAS PubMed.
- X. Chen, Y. Jiang, J. Sun, C. Jin and Z. Zhang, J. Power Sources, 2014, 267, 212–218 CrossRef CAS PubMed.
- T. Kou, D. Li, C. Zhang, Z. Zhang and H. Yang, J. Mol. Catal. A: Chem., 2014, 382, 55–63 CrossRef CAS PubMed.
- C. Zhang, X. Wang, J. Sun, T. Kou and Z. Zhang, CrystEngComm, 2013, 15, 3965–3973 RSC.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- D. P. Dubal, G. S. Gund, R. Holze and C. D. Lokhande, J. Power Sources, 2013, 242, 687–698 CrossRef CAS PubMed.
- Y. Li, G. Duan, G. Liu and W. Cai, Chem. Soc. Rev., 2013, 42, 3614–3627 RSC.
- Y. Li, N. Koshizaki, H. Wang and Y. Shimizu, ACS Nano, 2011, 5, 9403–9412 CrossRef CAS PubMed.
- X. Wang, Y. Yin, X. Li and Z. You, J. Power Sources, 2014, 252, 64–72 CrossRef CAS PubMed.
- H. B. Li, M. H. Yu, F. X. Wang, P. Liu, Y. Liang, J. Xiao, C. X. Wang, Y. X. Tong and G. W. Yang, Nat. Commun., 2013, 4, 1894 CrossRef CAS PubMed.
- H. Zheng, T. Zhai, M. Yu, S. Xie, C. Liang, W. Zhao, S. C. I. Wang, Z. Zhang and X. Lu, J. Mater. Chem. C, 2013, 1, 225–229 RSC.
- X. Zhang, W. Shi, J. Zhu, D. J. Kharistal, W. Zhao, B. S. Lalia, H. H. Hng and Q. Yan, ACS Nano, 2011, 5, 2013–2019 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |