DOI:
10.1039/C4RA11479E
(Paper)
RSC Adv., 2015,
5, 16265-16276
PEG based random copolymer micelles as drug carriers: the effect of hydrophobe content on drug solubilization and cytotoxicity†
Received
29th September 2014
, Accepted 12th January 2015
First published on 15th January 2015
Abstract
Polymeric micelles (PMs) of block copolymers have been extensively used as drug delivery vehicles in recent times. Polyethylene glycol (PEG) is being used for surface modification of these delivery systems, as PEG helps to protect from recognition by the reticuloendothelial system in the body. This work reports the synthesis and characterization of three new PEG-based random copolymers of poly(dodecyl methacrylate-co-polyethylene glycol methyl ether methacrylate), poly[DMAx-co-mPEGy] (where x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :y = 1:2, 1:4, 1:6), with different hydrophobe (DMA) content. These copolymers were observed to exhibit good surface activity in aqueous solution. The micelle formation by these random amphiphilic copolymers in aqueous medium was studied using a steady-state fluorescence probe technique. The size and shape of the PMs were determined by use of light scattering and electron microscopic techniques, respectively. The aqueous solutions of all three copolymers exhibit a temperature-induced phase transition above a critical temperature (LCST) forming a stable emulsion. The LCST value of the micellar solution was observed to increase with an increase of mPEG content of the copolymer, but decrease with an increase of polymer concentration. The PMs were successfully used to solubilize the hydrophobic anticancer drug (S)-(+)-camptothecin (CPT); the solubilization capacity of the PMs was observed to increase with the decrease of hydrophobe content (or with the substantial increase of hydrophile content) in these copolymers. However, all three copolymers have the potential to protect the labile lactone ring of CPT from hydrolysis. Though all three copolymers showed a considerable release of the guest at acidic pH, the polymer having the maximum hydrophile content showed maximum guest release. Furthermore, the PMs of all three copolymers were observed to be easily permeable to the cancer cell membrane. In the permissible concentration regime, the polymers were found to be nontoxic and hemocompatible, but the cell viability as well as hemocompatibility was observed to deteriorate with the increase of DMA content in the copolymers.
:y = 1:2, 1:4, 1:6), with different hydrophobe (DMA) content. These copolymers were observed to exhibit good surface activity in aqueous solution. The micelle formation by these random amphiphilic copolymers in aqueous medium was studied using a steady-state fluorescence probe technique. The size and shape of the PMs were determined by use of light scattering and electron microscopic techniques, respectively. The aqueous solutions of all three copolymers exhibit a temperature-induced phase transition above a critical temperature (LCST) forming a stable emulsion. The LCST value of the micellar solution was observed to increase with an increase of mPEG content of the copolymer, but decrease with an increase of polymer concentration. The PMs were successfully used to solubilize the hydrophobic anticancer drug (S)-(+)-camptothecin (CPT); the solubilization capacity of the PMs was observed to increase with the decrease of hydrophobe content (or with the substantial increase of hydrophile content) in these copolymers. However, all three copolymers have the potential to protect the labile lactone ring of CPT from hydrolysis. Though all three copolymers showed a considerable release of the guest at acidic pH, the polymer having the maximum hydrophile content showed maximum guest release. Furthermore, the PMs of all three copolymers were observed to be easily permeable to the cancer cell membrane. In the permissible concentration regime, the polymers were found to be nontoxic and hemocompatible, but the cell viability as well as hemocompatibility was observed to deteriorate with the increase of DMA content in the copolymers.
1. Introduction
Improvement of the therapeutic efficacy of drug molecules by developing a better delivery vehicle, rather than the drug molecule itself, is an emerging field in modern medication processes. Polymeric nanostructures (polymeric micelles,1–3 vesicles,4 nanoparticles,5 nanotubes,6,7 etc.) have drawn tremendous interest as drug delivery carriers due to their several advantageous properties like large surface area, various functionalities to the conjugate drug or receptor, stimuli sensitivity, easy synthesis and low cost. Polymeric micelles (PMs) were one of the first self-assembled nanostructures used in drug delivery in early 1990.1 Since then, PMs have been successfully used as promising drug delivery systems (DDSs) for chemotherapy because of their nano-range size, in vivo stability and structure being similar to those of some viruses and lipoproteins that are natural carriers in biological systems.8–10 Since PMs are composed of two distinct domains, a hydrophobic core and a hydrophilic corona, they can protect fragile molecules in their hydrophobic core from being destroyed by environmental conditions and allow them to remain unrecognized in circulation. Due to encapsulation within PMs, drug molecules can have a prolonged circulation lifetime and do not cause any embolization of blood vessels.11,12 This facilitates their extravasation at leaky capillaries, known as the “enhanced permeability and retention (EPR) effect”, and thereby leads to a passive accumulation in tumor tissues.13,14 Also with the increase of the molecular weight of the polymers/PMs, the in vivo stability is enhanced and as a consequence, the accumulation tendency at the tumor site gradually increases.15–17 Most of these polymers contain poly(ethylene glycol) (PEG) as the hydrophilic group, because PEGylation not only increases aqueous solubility and biocompatibility, but also it can prevent PMs from getting trapped by the reticuloendothelial system (RES), such as in the liver and spleen.17–20
As drug delivery vehicles, block copolymers21 as well as dendrimers22 rather than random copolymers have been investigated extensively due to their unique and excellent associating behavior with well-defined structures. But unlike block copolymers random copolymers can be synthesized very easily and their properties can also be regulated with an appropriate design of the copolymers.15,23–26 We, along with others, have shown that amphiphilic random copolymers have a strong tendency to form thermodynamically stable micelles in water above a very low critical aggregation concentration (CAC).14,27–31 The nature of aggregation (inter-polymer or intra-polymer) depends not only on the hydrophile–lipophile balance (HLB) but also on the linkage group (ester or amide), the nature of the hydrophobe and molecular weight.23,32 Therefore, exact choice and balance of the hydrophobe in the preparation of random amphiphilic copolymers is crucial to maintain and regulate their properties. In addition, this kind of polymeric nano-carrier with sensitivity to external stimuli has actually broadened their potential application ranging from drug delivery33,34 to personal care products35 to gene transfection.36 Depending on the nature of the stimulus (light,37,38 pH,39–42 temperature,39,41 redox reaction14 etc.), the encapsulated guest molecule can be triggered to the specific action site when it is needed. In the body, different organs, cells or tissues have a different pH. Therefore, pH can be a very attractive stimulus for drug delivery. Amphiphilic copolymers with a pH-sensitive/cleavable polyester backbone have been widely used for cancer chemotherapy, because after the release of the guest molecule the whole polymer can be degraded into small biodegradable fragments.
The objective of this study is to prepare, characterize, and evaluate pH-sensitive PMs with mPEG chains forming a hydrophilic shell as a part of a surface modification technique. Since PMs are highly effective43 for injectable DDSs, we have made an attempt to improve the aqueous solubility of the hydrophobic anticancer drug camptothecin (CPT) by use of PMs of random copolymers as an intravenous DDS. CPT is a potent anticancer drug, which generally exists in an active lactone form at pH values below 5, but converts to the inactive carboxylate form at basic pH (see Fig. 1(a)).44,45 At physiological pH, most CPT molecules, though poorly water-soluble, exist in an inactive carboxylate form. The lactone form also shows higher diffusibility through the lipid bilayer than the carboxylate form.46 In addition, CPT has a tendency to bind to human serum albumin (HSA) preferentially in the carboxylate form, resulting in a more rapid opening of the lactone ring.47 In order to increase the bioavailability and antitumor effects of CPT, DDSs must have the capability to protect the labile lactone ring as well as to increase the aqueous solubility. Keeping these facts in mind, we have designed and synthesized a new type of amphiphilic copolymer, poly(dodecyl methacrylate-co-polyethylene glycol methyl ether methacrylate), poly[DMAx-co-mPEGy] (Fig. 1(b)) with three different feed ratios [x:y = 1:2 (P12), 1:4 (P14), and 1:6 (P16)] of DMA and mPEG (Mn = 300). Although there are several PEG chains of various molecular weight with free –OH or –OMe groups, we have chosen the mPEG of Mn = 300 to see the properties of polymeric association when the length of the core- and corona-forming chains are similar. The choice of an –OMe group over an –OH group was based on two reasons: first, with the –OMe group there will be less chance of any non-specific interactions, and second, due to the presence of the –OMe group it would be easy to find a peak in 1H NMR spectra. The HLB was gradually changed in such a way that we can understand the effect of hydrophobe content on the various properties of the PMs. The PMs formed by these copolymers are expected to have a hydrophobic core made of DMA within which CPT can be entrapped, and a hydrophilic shell of PEG chains that will reduce interaction with HSA. The self-assembly of these copolymers was studied using surface tension, fluorescence, dynamic light scattering (DLS) and transmission electron microscopy (TEM) techniques. We have evaluated their potential as a DDS for CPT. We have also carried out in vitro evaluation of toxicity and blood compatibility of the copolymers. An attempt to demonstrate pH-triggered drug release has also been made.
 |
| | Fig. 1 (a) Chemical structure of the two forms of CPT; (b) chemical structure of poly[DMAx-co-mPEGy] copolymers with x:y = 1:2 (P12), 1:4 (P14) and 1:6 (P16). | |
2. Experimental section
2.1. Materials
Polyethylene glycol methyl ether methacrylate (mPEG; Mn = 300), dodecyl methacrylate (DMA), MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, a yellow tetrazole), chloroform-d, were purchased from Sigma-Aldrich (Bangalore, India) and were used without further purification. S-(+)-Camptothecin (CPT) was purchased from Tokyo Chemical Industry, Japan. 2,2′-Azo-bis-(isobutyronitrile) (AIBN) was purchased from Sigma-Aldrich (Bangalore, India) and was used after recrystallization from acetone. All the fluorescent probes like pyrene (Py), 1,6-diphenyl-1,3,5-hexatriene (DPH), N-phenyl-1-naphthyl amine (NPN) and coumarin 153 (C-153) were purchased from Sigma-Aldrich (Bangalore, India). Solvents like methanol (MeOH), dimethyl formamide (DMF), tetrahydrofuran (THF), ethyl acetate, petroleum ether, and chlorinated solvents were purchased from Merck and were distilled and purified before use. Milli Q (18.2 MΩ) water was obtained from a Millipore water purifier (Elix, Bangalore, India).
2.2. Synthesis of copolymers
The copolymers P12, P14 and P16 were synthesized according to Scheme S1 of the ESI† using different mole ratios of the hydrophobic monomer, DMA, and hydrophilic monomer, mPEG, by a reversible addition–fragmentation chain transfer (RAFT) polymerization technique. The chain transfer agent (CTA) 2-(2-cyanopropyl)-dithiobenzoate (CPDB, Fig. S1†) was employed as there are already many reports for successful utilization of CPDB for polymerization of acrylate based monomers.48,49 The details of the synthesis and chemical identification of CPDB are included in the ESI.† The reaction medium of a dry DMF–THF mixture (4:1, v/v) was purged with N2 gas for 30 minutes and after that the CTA (1/500 mole ratio with respect to total reactant) and AIBN (1/5 mole ratio with respect to CTA) were added and then again purged with N2 gas for 30 minutes, then finally the reaction mass was heated at 70–75 °C for 46–48 h. The reaction was quenched by keeping the flask in chilled water. Then the solvents were evaporated under high vacuum to concentrate the reaction mass and water was added to it. The clear water part was taken for dialysis against distilled water for 4–5 days with intermittent change of water. Water was then removed by a lyophilization technique and the polymer was obtained as a viscous liquid. The chemical structures of the copolymers were confirmed by 1H- and 13C-NMR, and Fourier transform infrared (FT-IR) spectra. Representative 1H-NMR spectra of the copolymer are presented in Fig. S2–S4 in the ESI.†
2.3. Molecular weight determination
The average molecular weight (![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) w) and polydispersity index (PDI) of the copolymers were determined by gel permeation chromatography (GPC, Waters 2414, Refractive Index Detector, Waters 515 HPLC PUMP) using poly(methyl methacrylate) (PMMA) as the molecular weight standard. DMF (HPLC grade) was employed as an eluent at a flow rate of 0.3 mL min−1 at 25 °C.
w) and polydispersity index (PDI) of the copolymers were determined by gel permeation chromatography (GPC, Waters 2414, Refractive Index Detector, Waters 515 HPLC PUMP) using poly(methyl methacrylate) (PMMA) as the molecular weight standard. DMF (HPLC grade) was employed as an eluent at a flow rate of 0.3 mL min−1 at 25 °C.
2.4. General instrumentation
1H and 13C-NMR spectra were recorded on an AVANCE DAX-400 (Bruker, Sweden) 400 MHz NMR spectrometer using TMS as the internal standard. The FTIR spectra were recorded on a Perkin-Elmer (model 883 IR) spectrometer. Electronic spectra were recorded by use of a UV-vis spectrophotometer (Shimadzu, model UV-2450). For solid samples, KBr pellets were used as solvent. The solution pH was measured using a digital pH meter model 5652 (EC India Ltd., Calcutta) using a glass electrode.
2.5. Surface tension (ST) measurement
A du Nuöy ring detachment method was employed for surface tension (γ) measurements of the aqueous copolymer solutions at 25 °C using a semiautomatic surface tensiometer (model 3S, GBX, France). The platinum ring was used after appropriate cleaning with ethanol–HCl (1:1, v/v) solution and burning in an oxidizing flame immediately before every use. For each polymer, the γ value of water was measured first and then an aliquot from the stock polymer solution was added to it gradually for the surface tension measurements at different polymer concentrations. Before measurement, each sample was equilibrated for 15–20 minutes. For each concentration, the γ value was measured in triplicate and the mean value was noted.
2.6. Fluorescence measurements
Steady-state fluorescence. The microenvironment of the PMs in water was characterized using steady-state fluorescence spectroscopy using Py, DPH and NPN as the fluorescent probes. A SPEX Fluorolog-3 spectrophotometer (450 WATT ILLUMINATOR, Model: FL-1039/40, HORIBA JOBIN YVON, EDISON, NJ, USA) was used for the steady-state fluorescence measurements. The CAC of the polymers in aqueous media was measured using a Py probe. Aliquots of Py stock solution (1.0 × 10−3 M in MeOH) were added into 5 mL volumetric flasks and the solvent was evaporated by a continuous stream of N2 gas. Then, polymer solutions in Milli-Q water in different concentrations were added to the volumetric flasks, making the final concentration of Py 1.0 × 10−6 M. Before measurement, more than 12 h of incubation time was given to all the polymer solutions. The solutions containing the Py probe were excited at 343 nm, and the emission spectra were recorded in the range of 350–600 nm. The excitation and emission slit widths were 5 and 2 nm, respectively.The microviscosity and micropolarity of the PMs were estimated using DPH and NPN probes, respectively. Solutions containing NPN (ca. 1 × 10−6 M) were excited at 340 nm and emission spectra were recorded in the wavelength range 350–550 nm on the SPEX Fluorolog-3 spectrophotometer. Both excitation and emission slit widths were fixed at 5 nm. DPH was used as a fluorescence anisotropy probe. Steady-state fluorescence anisotropy (r) of DPH was measured on a Perkin-Elmer LS-55 spectrophotometer equipped with a thermostating cell holder and filter polarizer/analyzer assembly that used the L-format configuration. For anisotropy measurements excitation and emission slit widths were fixed at 2.5 nm and 5 nm, respectively. Each anisotropy measurement was repeated at least six times and the mean value of r was noted. The temperature of the samples was controlled using the water jacketed magnetically stirred cell holder in the spectrometer connected to a Thermo Neslab RTE-7 circulating water bath that enabled temperature control within ±0.1 °C.
Time-resolved fluorescence. Fluorescence lifetime of the DPH probe in the polymer solution was measured by a time-correlated single-photon counting (TCSPC) technique using a nano-second diode laser at λ = 380 nm (EasyLife) as the light source. The fluorescence decay kinetics of DPH were recorded at the emission wavelength of 450 nm. The data analysis was performed using the software provided by the supplier. The decay curves were fitted to a double exponential decay function. The goodness of the fit was judged by the randomness of the residual plot as well as by the χ2 value (0.8–1.2).
2.7. Dynamic light scattering (DLS)
The hydrodynamic diameter (dH) of the PMs in aqueous media was measured by the conventional DLS technique using a Malvern Nano ZS instrument employing a 4 mW He–Ne laser (λ = 632.8 nm). All the scattering photons were collected at a 173° scattering angle. The temperature was set to 25 °C and before every measurement each polymer solution was filtered through 0.45 μm (Millipore Millex syringe filter) filter paper. The software provided by the supplier calculated the hydrodynamic size using the Stokes–Einstein equation.
2.8. Transmission electron microscopy (TEM)
A high resolution transmission electron microscope (HRTEM, TECNAI G2-20S TWIN, Japan) operating at an accelerating voltage of 200 kV at room temperature was employed to take micrographs of the polymers solutions at a known concentration above their CAC value. A 5 μL aqueous polymer solution was drop cast on a carbon-coated copper grid (400 mesh size) and it was kept in vacuum desiccators overnight for drying.
2.9. Solubilization study
A solvent evaporation method33,50 was used to solubilize CPT. Exactly the same amount of the drug with a different amount of polymer (required for various concentrations) was dissolved in 2 mL of CHCl3 and sonicated for 5 min in a bath -type sonicator (Bandelin, Sonorex, ultrasonic frequency 35 kHz). Then the CHCl3 solution was added drop wise into 10 mL of Milli Q water under vigorous stirring conditions. After complete evaporation of CHCl3 from this heterogeneous mixture by simple stirring, the solution was left stirring for another 24 h in the dark to ensure the maximum solubilization of CPT. The supernatant was then filtered using a Millipore Millex filter (0.45 μm pore diameter) to remove the insoluble drug, if any. A measured volume of the filtrate was diluted with MeOH to measure the absorbance value at a wavelength of 360 nm using the previously recorded calibration curve. The copolymer solutions without any drug at the same dilution were used as a reference.
2.10. Hemolytic assay
Approximately 5 mL of human blood was freshly collected from a known healthy volunteer with consent just before the experiment. All experiments involving blood samples were performed in compliance with the relevant laws and guidelines of the "Institute Ethical Committee", Indian Institute of Technology Kharagpur. A hemolytic assay was performed following the standard protocol.51 Briefly, red blood cells (RBCs) were collected from the human blood by centrifugation at 3000 rpm for 10 min at room temperature and were washed 4 times with 150 mM NaCl solution followed by a suspension in PBS (pH 7.4) to a final cell concentration of 5 × 108 RBC per mL. For making the 1 mL sample solution, different amounts of polymer (in PBS, pH 7.4) were added into 200 μL of the above RBC suspension and the final volume was made up by PBS. As positive and negative controls, RBCs were suspended in triton X-100 (1%, w/v) and PBS, respectively. All the samples were incubated for 60 min at 37 °C with intermittent mixing followed by centrifugation at 12000 rpm for 5 min. The absorbance of the supernatant solutions was taken at 541 nm using an ELISA reader (Biorad, USA) with PBS as the blank. Each experiment was performed in triplicate.
2.11. Cell viability assay
The cell viability assay was performed following the standard protocol with some modifications.50 HeLa cells (cervical cancer cell line) and L929 cells (normal cell line) were cultured in DMEM (Dulbecco’s Modified Eagle’s Medium) supplemented with 10% fetal bovine serum (FBS), and an antibiotic solution containing penicillin (100 units per mL), streptomycin (0.1 mg mL−1), and amphotericin B (0.25 μg mL−1). The cells were maintained at 37 °C in T-25 flasks in a humidified incubator (5% CO2) with a feeding cycle of 48 h. The confluent cell monolayer was trypsinized (0.25% Trypsin + 0.1% EDTA) and the cells were harvested by centrifugation.
In an incomplete DMEM media (pH 7.4), the copolymers were dissolved and then filtered through a 0.2 μm polycarbonate filter after 12 h of incubation. Cell suspensions were seeded at 3 × 103 cells per well in 100 μL of complete DMEM in a 96-well plate. The cells were allowed to adhere and were grown for 24 h at 37 °C in an incubator. The medium was aspirated and replaced with 100 μL of fresh medium containing the control and polymer at the desired concentrations. After 24 h of incubation with the polymers, the medium was removed and the cells were washed thrice with sterile PBS. Cell viability was performed using a conventional MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) dye reduction assay. 100 μL of MTT reagent (5 mg mL−1 in PBS) was added to each well and incubated for 3 h. The MTT reagent mixture was gently removed and 200 μL of DMSO was added into each well. The developed formazan dye was measured spectrophotometrically at 540 nm. This experiment was performed in triplicate. The cell viability for these polymers was expressed as a percentage with respect to the untreated control cells. The following formula was applied to calculate cell viability.
| Cell viability (%) = (mean of absorbance value of treated cells/mean of absorbance value of untreated control cells) × 100. |
2.12. Cellular uptake
Confocal fluorescence microscopic images were taken using C-153 as the fluorescent probe (excitation at 422 nm and emission at 510 nm) to confirm whether these polymers can cross the cell membrane.52 A stock solution (1.0 × 10−3 M) of C-153 was prepared in MeOH and an aliquot was added into 5 mL volumetric flasks so that the final concentration of C-153 became ca. 1.0 × 10−6 M. Then MeOH was evaporated by a stream of N2 gas and polymer solutions (in PBS, pH 7.4) of different concentrations were prepared in these volumetric flasks following the usual method. After 12 h of incubation in the dark at room temperature each sample was dialyzed against PBS solution (pH 7.4) for 2–3 h using a biodialyser to completely remove the insoluble dye. HeLa (cervical cancer cells) cells of mid log phase growth were seeded in a 24-well flat bottom plate at a concentration of 3 × 103 per well and were left to grow for 24 h at 37 °C in a CO2 environment. Then the culture media was replaced with the polymer solution containing C-153. After 3 h of incubation at 37 °C, the sample solution was aspirated and the wells were washed gently 3–4 times with PBS and were finally suspended in 200 μL of PBS before taking the images using an Olympus confocal microscope (FV1000, Olympus).
3. Results and discussion
3.1. Molecular characterization of copolymers
Polymerization was confirmed by the disappearance of vinylic proton signals in the1H-NMR spectrum (Fig. S2–S4, ESI†). The DMA/mPEG mole ratio in each copolymer was determined using the respective 1H-NMR spectrum. The average molecular weights (w) of the polymers, P12, P14, and P16, as obtained from GPC were 35279, 36728, and 45611, respectively. The relatively low PDI values of P12 (1.26), P14 (1.31), and P16 (1.37) suggest a narrow molecular weight distribution for each copolymer. The chromatograph (Fig. S5, ESI†) clearly reveals that all three polymers have molecular weights in a very close range in spite of having different DMA/mPEG mole ratios. As observed, the molecular weight increased with the increase of overall mPEG content in the polymer backbone and with a gradual increase of the PDI value. Thus the copolymer P16 has the highest average molecular weight among all the polymers.
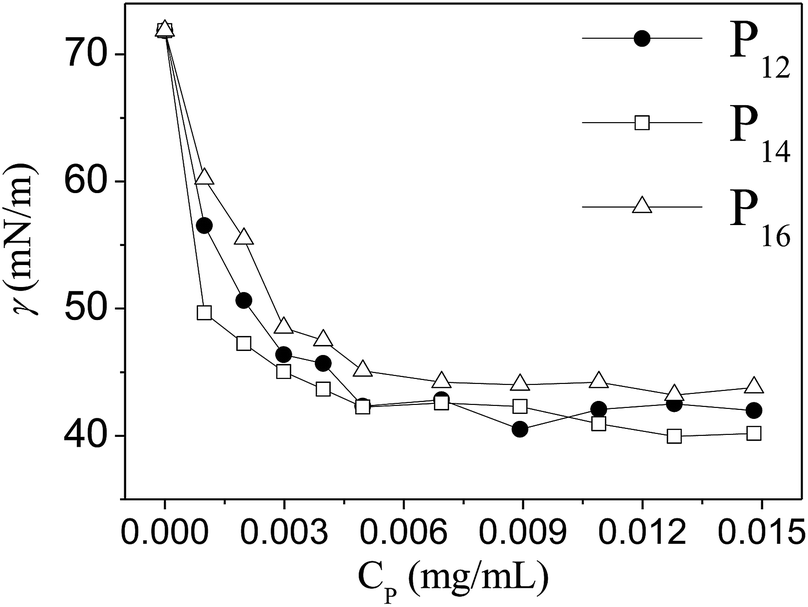
3.2. Surface activity
All three copolymers are expected to behave like surface-active agents as the polymer backbone contains both hydrophilic and hydrophobic groups. Therefore, we measured the surface tension (γ) of water in the presence of various concentrations (CP) of the copolymers at room temperature. The data are plotted in Fig. 2. It is evident that all three polymers reduced the γ-value of water to ca. 45 mN m−1 at a very low concentration, showing good surface activity. The data in Fig. 2 suggest that the copolymer (P16) having the lowest hydrophobe (DMA) content is least surface active. Such a large reduction of the surface tension of water proved the amphiphilic nature of all the copolymers. The polymer concentration (CP ∼ 7 × 10−3 mg mL−1) corresponding to the minimum γ-value can be taken as the CAC value of the copolymer. Thus all the polymers have a very low CAC value.
 |
| | Fig. 2 Variation of surface tension (γ) of water with the concentration (Cp, mg mL−1) of copolymers, P12, P14 and P16. | |
3.3. Self-assembly formation
All the copolymers are expected to self-assemble in water at a concentration greater than the CAC (ca. 7 × 10−3 mg mL−1) as they behave like amphiphilic molecules. The self-assembly formation of the copolymers was studied using a steady-state fluorescence technique using NPN, pyrene, and DPH as probe molecules. These hydrophobic fluorescent probes are known to be solubilized within the hydrophobic core of the (micellar) aggregates.53,54 Both NPN and DPH are weakly fluorescent in water, but the fluorescence intensity increases in a non-polar medium or within the hydrophobic core of the micelles.54 The fluorescence spectrum of NPN in the presence of polymer exhibits a large blue shift (Fig. S6†) in addition to an intensity rise (Fig. 3(a)), indicating the existence of hydrophobic domains, the formation of which is concentration dependent and therefore can be attributed to micelles.54 For determination of the CAC values of the copolymers, the relative fluorescence intensities (F/Fo, where F and Fo are respectively the fluorescence intensities in the presence and absence of polymer) of NPN at different polymer concentrations were measured. The data are plotted against Cp in Fig. 3(b). The plots show that for all three copolymers, F/Fo is very low and is independent of Cp in dilute solutions, but they exhibit a sharp rise above a critical concentration. This observation leads to the conclusion that the aggregation started above this critical concentration through the inter-chain aggregation and no unimer micelles are formed. The Cp value corresponding to the onset of intensity rise was taken as the CAC value. It is observed that the CAC values of all three polymers are nearly equal to 5 × 10−3 mg mL−1, close to that obtained from the ST method.
 |
| | Fig. 3 (a) Fluorescence spectra of NPN in pure water and in the presence of copolymers (Cp = 0.02 mg mL−1) at 25 °C; (b) plots of the relative fluorescence intensity (F/Fo) of NPN versus polymer concentration (Cp) in water at 25 °C. | |
3.4. Hydrodynamic size of the PMs
The hydrodynamic size of the PMs is an important parameter for drug delivery applications, because nanosized micelles have a lower tendency for rapid clearance from circulation by phagocytosis in post intravenous administration than microparticles, and also nanoparticles show an efficient EPR effect and minimal renal excretion for passive tumor targeting.55 As blood capillaries in the body are 5–6 μm in diameter, the size of the particles administered into the bloodstream must be significantly smaller than 5 μm without the possibility of embolism. A size less than 100 nm with a modified hydrophilic surface is crucial for achieving the reduction of opsonization reactions.56 Therefore, the hydrodynamic diameters of the PMs in aqueous solutions (1.0 mg mL−1) of the copolymers were measured using a DLS technique. The size distribution histograms are shown in Fig. 4(a–c). It is observed that both polymer P12 and P14 show broad distributions with mean hydrodynamic diameters of ca. 65–80 nm in comparison to P16, the PMs of which have a mean hydrodynamic diameter of ca. 100 nm. This is consistent with the higher molecular weight of P16. We also performed DLS measurements at various concentrations of all the three copolymers. The results are presented in Fig. S7(a–c) in the ESI.† The data suggest that the size distribution profiles become sharper with the increase of polymer concentration. Thus the PMs formed by these amphiphilic polymers are in an acceptable range for drug delivery applications.
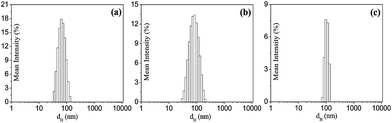 |
| | Fig. 4 Hydrodynamic size distribution of the aggregates formed in aqueous solutions (1.0 mg mL−1) of the copolymers (a) P12, (b) P14 and (c) P16 at 25 °C. | |
3.5. Shape of the PMs
The shape of the PMs was visualized using TEM. The micrographs (Fig. 5(a–c)) of the copolymer solutions (1.0 mg mL−1) clearly reveal the presence of spherical particles (micelles) with diameters in the range of 50–100 nm and a reasonably narrow size distribution. These results are similar to those obtained from DLS measurements.
 |
| | Fig. 5 TEM images of the PMs in aqueous solution (1.0 mg mL−1) of (a) P12, (b) P14 and (c) P16 at 25 °C. | |
3.6. Micropolarity of the PMs
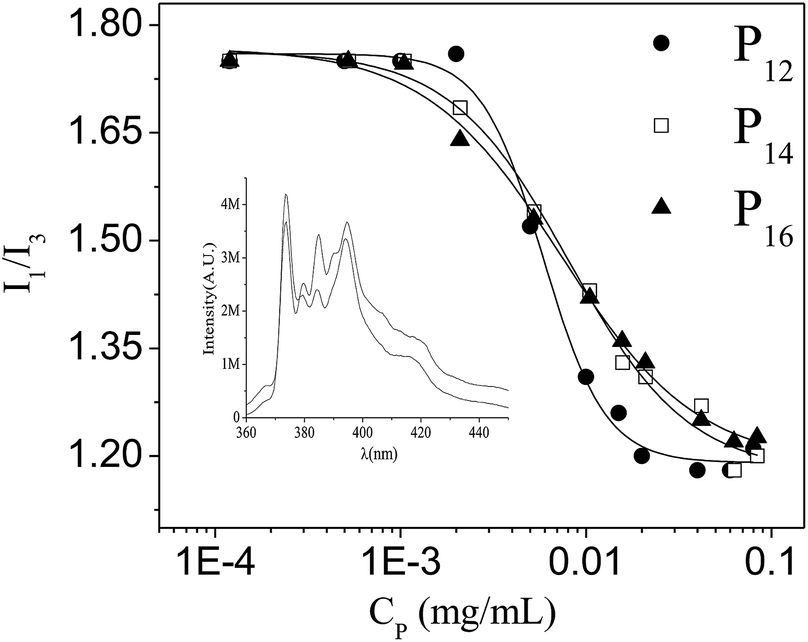
In the Py emission spectrum among the five vibronic peaks, the intensity ratio of the first and the third vibronic peak (I1/I3) is known to be very sensitive to the polarity of the medium and therefore is used as an index of solvent polarity.57,58 The fluorescence spectra of Py were recorded in the presence of different concentrations of each of the polymers. The I1/I3 ratio measured at different polymer concentrations is plotted in Fig. 6. Representative spectra are shown as the inset of Fig. 6. One can easily see that below a critical concentration (equal to the CAC), the I1/I3 ratio is close to the value in pure water (1.8), but it decreases non-linearly above the CAC with the increase in polymer concentration, reaching a minimum at about 0.1 mg mL−1. The CAC values (4 × 10−3–7 × 10−3 mg mL−1) as obtained from the inflection point of the plots are similar to the values obtained from fluorescence titrations using a NPN probe. Thus the accuracy of the determined CAC values of the polymeric amphiphiles in aqueous media is further confirmed. The initial concentration-independent plateau at low concentrations confirms inter-molecular association of the polymers. In general, in this type of amphiphilic random copolymer there is always a tendency to form intra-polymer aggregates.59 But because of low PDI values (i.e., greater homogeneity of macromolecules) and probably not having an appropriate w and enough hydrophobe content in the polymer backbone, only inter-polymer association occurs.23,59 It should also be noted that ester linkage always helps the formation of inter-polymer aggregates.23 The minimum value of the I1/I3 ratio (∼1.2) of Py, for all the polymers, indicates that the polarity of the microenvironment is similar to THF-like organic solvents.53
 |
| | Fig. 6 Plots of the intensity ratio (I1/I3) of Py fluorescence spectra versus copolymer concentration (Cp). Inset: Fluorescence emission spectra of Py showing I1 and I3 bands in water and in the presence of polymer. | |
3.7. Microviscosity of the PMs
Steady-state fluorescence anisotropy (r) of the DPH probe was measured in the presence of different concentrations of the polymers in order to determine the rigidity of the microenvironment of the PMs. The plots of variation of r with the Cp are depicted in Fig. S8 (ESI),† which shows that the r-value for the polymeric micelles of the copolymers is quite high, indicating a very rigid (i.e. viscous) microenvironment.60,61 This is expected because the long fatty acid chains, that are covalently linked to the polymer backbone, make the polymer less mobile. This may be the reason why the copolymer with a comparatively higher hydrophobe content (P12) showed a higher r-value at the same polymer concentration. The microviscosity (ηm) of the PMs was determined using the r-value and fluorescence lifetime data of the DPH probe according to the Stokes–Einstein–Debye (SED) equation:62| | |
ηm = kBTτf/[vh(ro/r − 1)]
| (1) |
where τf is the fluorescence lifetime of the DPH probe, vh is the hydrodynamic volume of the DPH (313 Å3) molecule, ro (= 0.362) and r are the fluorescence anisotropy values of DPH in a highly viscous medium and in the presence of polymer respectively, kB is the Boltzmann constant, and T is absolute temperature. The data are collated in Table S1 (ESI†). The ηm values for the copolymers P12 (175 mPa s), P14 (141 mPa s), and P16 (104 mPa s) were observed to be quite high in comparison to micelles of neutral surfactants, e.g. Triton-X100 (39.81 mPa s).62 The data also show that as the PEG-content increases the ηm value decreases. This is because of the lack of hydrophobic interaction among the DMA chains forming the core of the polymeric micelles.
3.8. Solubilization of hydrophobic drugs
The I1/I3 and ηm data of the PMs of the P12, P14 and P16 copolymers suggest that they are capable of solubilizing lipophilic molecules within the hydrophobic core. Therefore, we studied the aqueous solubility of the poorly water-soluble drug CPT in the presence of different concentrations of the copolymers at room temperature. The results are presented in Fig. 7. The plots in Fig. 7 show that the solubility of CPT in water increases with the gradual increase of polymer concentration due to physical entrapment of the drug within the hydrophobic core of the PMs. Although all the polymers can be used as a good sequestering agent for CPT, P16 showed the highest drug loading (29.6 mg g−1 or 2.96% w/w) in comparison to those of P14 (23.2 mg g−1 or ca. 2.32% w/w) and P12 (20 mg g−1 or 2.0% w/w). This is surprising because according to common belief, PMs of polymers with higher hydrophobe content should have low micropolarity and consequently, they should solubilize hydrophobic drugs more. The lower solubility of CPT in the micelles of P12 and P14 in comparison to P16 is due to their low permeability. Also in comparison to P14 and P12 the molecular weight of P16 is quite high. However, the drug loading capacity and entrapment efficiency (Table S2, ESI†) of the PMs of the copolymer are reasonably good. However, the drug loading capacity of these copolymers is better than those of the polymer–drug conjugates. Indeed, the CPT loading by its conjugate to a PEG with a molecular weight of 40 kDa was only 0.86–1.72% (w/w).63,64 So, these PMs can be used easily for passive targeting of any kind of hydrophobic drug. It should also be noted that the fluorescence spectra of the CPT molecule (Fig. S9a–c, ESI†) when solubilized within the PMs match with the fluorescence spectrum of its lactone form, suggesting solubilization of the active form of the drug molecule.65 Furthermore, it is observed that the intensity of the fluorescence spectra of CPT-loaded PMs decreased with the increase of polymer concentration. This occurrence of concentration quenching of CPT fluorescence also strongly supports solubilization of more than one CPT molecule within each PM. The fluorescence spectra of CPT at various pH and in the PMs of P12, P14 and P16 are presented in Fig. S9a–d (ESI†). Under basic conditions (pH 12), the λmax of CPT fluorescence exhibited a red shift accompanied by the decrease of intensity in comparison to that at pH 7, indicating hydrolysis of the lactone ring.45 At pH 7, in the presence of polymeric micelles, however, the spectrum remained unchanged over a long period of time showing protection of the hydrolytically labile lactone ring.
 |
| | Fig. 7 Plots of solubility (μg mL−1) of CPT drug versus copolymer concentration (Cp) in water at 25 °C. | |
3.9. Thermal stability of the PMs
PEG-containing polymers are known to exhibit a lower critical solution temperature (LCST) due to temperature-induced dehydration of the PEG chain (as a result of the rupture of the H-bonding between PEG and water).66 Therefore we monitored the percentage transmittance (%T) of the aqueous solutions of the polymers with Cp > CAC at different temperatures. The plot %T versus temperature for each of the polymer solutions with Cp = 1.0 mg mL−1 is shown in Fig. 8. As can be seen, the transmittance falls sharply with the rise of temperature in all the cases. This is due to the appearance of turbidity which increases with the increase of temperature. The appearance of turbidity must be caused by the dehydration of the PEG chain at elevated temperatures which is a common feature of many PEG-containing block copolymers.66 The PMs with dehydrated PEG chains on the surface transform into nanosized oil droplets. The phenomenon was observed to be reversible, that is, the solution becomes clear when cooled back to room temperature. The phase transition temperature can be taken as the LCST. The LCST value was observed to increase in the order P12 (46 °C) < P14 (50 °C) < P16 (55 °C), i.e., in the order of increasing PEG content of the polymer. Similar experiments (Fig. S10, ESI†) using a lower concentration (Cp = 0.5 mg mL−1) of the polymers, however, resulted in a slightly blue color dispersion. Interestingly, for any given polymer, the LCST value was observed to be higher than that of the corresponding value in 1.0 mg mL−1 polymer solution. However, the LCST value of the 0.5 mg mL−1 polymer solutions increased in the same order (P12 (55 °C) < P14 (60 °C) < P16 (66 °C)). The appearance of turbidity at higher temperatures occurs as a result of coalescence of the nanosized oil droplets to form larger droplets leading to the formation of a stable O/W emulsion. The formation of larger sized particles on heating of the polymer solution was confirmed by the results of DLS measurements. As can be seen in Fig. S11(a–c) (ESI†), with all three polymers, the size distribution histograms become broader showing a gradual increase of hydrodynamic diameter with the increase of temperature.
 |
| | Fig. 8 Plots of transmittance (%) as a function of temperature (°C) for aqueous polymer solutions (1.0 mg mL−1); Inset: photographs of the vials containing polymer solutions (a) below and (b) above the LCST. | |
It is important to note that even at a higher concentration, the LCST values of the polymers are well above the physiological temperature (37 °C). Since the LCST values of all three copolymer solutions are well above 37 °C at this high concentration (Cp = 1.0 mg mL−1), the clouding of the solution will not occur at body temperature. Consequently, premature release of any encapsulated pharmaceutically active agent at body temperature can be ruled out.
3.10. pH-stability of the PMs
Since the hydrophobes (DMA) and hydrophiles (mPEG) are linked to the polymer backbone through ester linkages, the PMs can be easily hydrolyzed in acidic or alkaline medium. Thus depending on the pH change, the encapsulated guest within the microenvironments of the PMs can be released. Since tumor cells have a pH in the range 4.5–6.5,33,39–41 the stability of the PMs at acidic pH (pH 4.7) was measured by using the intensity change of DPH fluorescence at the physiological temperature (37 °C). For these measurements, all the polymers were employed at a Cp value equal to 1.0 mg mL−1, as the encapsulation efficiency of the PMs was observed to be highest at this concentration. These results are presented in Fig. 9. Due to the hydrolysis of the ester bonds in the polymer branches, the fluorescence intensity of the DPH probe decreases, indicating a gradual release of DPH molecules from the PMs. In almost a 2 h period, 50% of the encapsulated guest molecule is released following a first order decay rate for P12 and P14, whereas about 70% release was observed with P16. The higher amount of release of DPH in the case of P16 is consistent with its high mPEG content which makes the PMs more permeable compared to those of the P12 and P14 polymers. As a control experiment, the release study at pH 7.4 was also performed, but no significant release of DPH could be observed within the period of measurement. This clearly suggests that the release of DPH is due to the hydrolysis of the ester bonds connecting DMA and PEG chains to the polymer backbone. To further confirm hydrolytic cleavage of the DMA and PEG chains, DLS measurements (Fig. S12(a–c), ESI†) of the aqueous solution of P12, P14 and P16 at pH 4.7 and 1.0 were performed after ∼14 h of incubation at 37 °C. The size distribution profiles clearly show formation of smaller sized PMs along with larger sized particles due to the polymethacrylic acid, indicating degradation of the mPEG as well as the DMA chains from the polymeric backbone.
 |
| | Fig. 9 Cumulative (%) release of DPH as a function of time (min) at pH 4.7 and 37 °C. | |
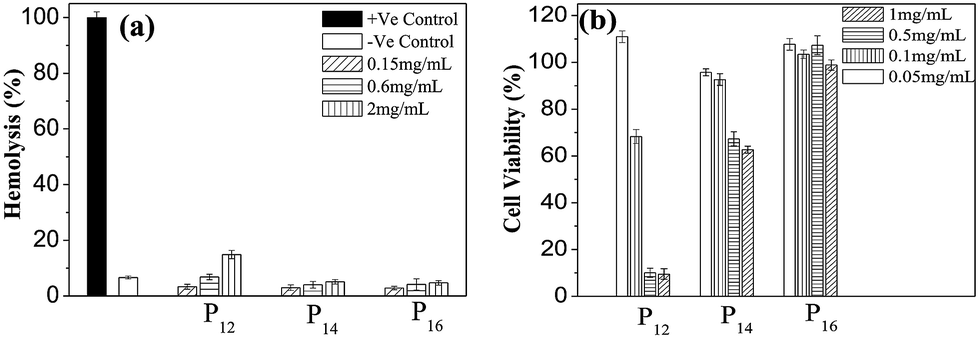
3.11. Hemocompatibility of the copolymers
Hemocompatibility of the polymers is a necessary requirement for intravenous (i.v.) drug delivery systems. A hemocompatibility assay was performed by studying the interaction of collected human RBCs with the copolymers. The percentage of hemolysis of RBCs at various concentrations (0.15 to 2.0 mg mL−1) of copolymers is plotted in Fig. 10(a) and compared with the negative (RBCs suspended in PBS) as well as positive (RBCs suspended in 1% triton X-100) controls. Interestingly, all the polymers were found to be hemocompatible at moderate to high concentrations. However, at elevated concentrations (2.0 mg mL−1), P12 clearly shows the highest hemolysis (though still very low) among all the polymers due to its highest hydrophobe content.50 The other two polymers, however, have shown more or less the same hemocompatibility at all concentrations employed. The results indicate that perhaps the P14 and P16 polymers have potential uses as intravenous DDSs.
 |
| | Fig. 10 (a) Hemolysis assay of copolymers (0.15 to 2.0 mg mL−1) at physiological pH (7.4); (b) cytotoxicity effects of the copolymers (0.05 to 1.0 mg mL−1) on HeLa cells based on an MTT assay after 24 h of incubation with respect to untreated control cells. Cell viability without polymer (control) was considered as 100%. The data are presented as the mean ± SD. | |
3.12. In vitro cytotoxicity assay
For the cell viability assay of the polymers, both cancer (HeLa) and normal (L929) cells were cultured and treated with different concentrations (0.05–1.0 mg mL−1) of the polymers followed by the detection of the cytotoxicity using an MTT assay. As observed from Fig. 10(b), for P16, cell viability remains very high even at very high concentrations (0.5 and 1.0 mg mL−1). Though P14 is still compatible at this concentration, P12 is clearly cytotoxic. The results clearly suggest that with the increase of mPEG content in the polymer backbone the cell viability gradually increases, and as a result, P16 is the least toxic among the three polymers. The same trend of the hydrophilic effect on cell viability can also be observed with the normal cells (L929) (Fig. S13, ESI†).
3.13. In vitro cellular uptake of PMs
The confocal microscopy images showed a bright blue fluorescence (Fig. 11(a)) when the HeLa cells were treated with C-153 encapsulated PMs of the different polymers (CP 0.25 mg mL−1). This blue fluorescence indicated that the cancer (HeLa) cells can easily uptake these PMs in their cellular compartment within a short incubation time of 3 h at 37 °C. However, the cellular compartmentalization was found to be independent of the mPEG content of the copolymers. Confocal micrographs of cell sections (Fig. 11(b)) are also helpful to understand the intracellular localization of the probe.
 |
| | Fig. 11 (a) Confocal micrographs of HeLa cells incubated with C-153 loaded PMs of the copolymers. Untreated cells were taken as control; scale bar is 60 μM. (b) Confocal micrographs of cell sections showing internalization of different polymers. Scale bar is 20 μM. | |
4. Conclusions
In summary, we have synthesized three mPEG based neutral amphiphilic random copolymers (P12, P14 and P16) with different hydrophobe (DMA)/hydrophile (mPEG) mole ratios using a RAFT copolymerization technique. All the copolymers were observed to form micelles in water above a very low critical concentration through inter-chain association. The polymeric micelles (PMs) thus formed were found to have a similar spherical morphology. The PMs have a mean hydrodynamic size in the range of 70–100 nm and have a less polar and relatively rigid microenvironment compared to nonionic surfactant micelles. As a result, the PMs of the copolymers were observed to solubilize and protect a poorly water-soluble anticancer drug, such as CPT, within the less polar microenvironment. The aqueous solubility of CPT increased 8–12 times in the presence of 1.0 mg mL−1 polymer. However, the solubilization capacity of these copolymer micelles was observed to be less than those of the corresponding cholesterol-containing copolymers.67 Like cholesterol-containing copolymers, all three copolymers also exhibit a LCST and the LCST value increased with the increase of mPEG content as well as with dilution. Above the LCST the PMs are transformed into partially dehydrated oil droplets thus producing a stable O/W emulsion. However, all three copolymers were observed to be quite stable and remain water-soluble at a concentration as high as 1.0 mg mL−1 at the physiological temperature and pH. On the other hand, in acidic pH, the polymers undergo hydrolytic cleavage disrupting the PMs. This triggered release of encapsulated DPH, a hydrophobic model drug. In vitro biological studies have shown that all three copolymer micelles can enter the cellular compartment and except P12, they do not exhibit any significant cytotoxicity at Cp ≤ 1.0 mg mL−1. The cell viability study suggested that the copolymers with a higher hydrophobe content are more toxic. However, cytotoxicity of these copolymer micelles was observed to be comparable to those of the corresponding cholesterol-containing copolymers.67 These copolymers were also found to be hemocompatible up to a relatively high concentration (2 mg mL−1). Based on these results it can be concluded that copolymers P14 and P16 can be used as intravenous delivery systems in the viable concentration range.
Acknowledgements
The authors gratefully acknowledge the Department of Science and Technology, New Delhi for the financial support (Grant no. SR/S1/PC-68/2008) of this work. PL thanks CSIR, New Delhi (no. 20-06/2010 (i) EU-IV) for a research fellowship. We sincerely acknowledge Dr S. Ghosh, Polymer chemistry division, IACS, Kolkata for GPC measurements. We thank Prof. Hideki Matsuoka, Kyoto University, JAPAN for providing CPT as a gift.
Notes and references
- A. S. Hoffman, The origins and evolution of “controlled” drug delivery systems, J. Controlled Release, 2008, 132(3), 153–163 CrossRef CAS PubMed.
- C. M. J. Hu, R. H. Fang, B. T. Luk and L. Zhang, Polymeric nanotherapeutics: clinical development and advances in stealth functionalization strategies, Nanoscale, 2014, 6(1), 65–75 RSC.
- S. Yu, C. He, J. Ding, Y. Cheng, W. Song, X. Zhuang and X. Chen, pH and reduction dual responsive polyurethane triblock copolymers for efficient intracellular drug delivery, Soft Matter, 2013, 9(9), 2637–2645 RSC.
- Z. Y. Qiao, R. Ji, X. N. Huang, F. S. Du, R. Zhang, D. H. Liang and Z. C. Li, Polymersomes from dual responsive block copolymers: Drug encapsulation by heating and acid-triggered release, Biomacromolecules, 2013, 14(5), 1555–1563 CrossRef CAS PubMed.
- B. Khorsand, G. Lapointe, C. Brett and J. K. Oh, Intracellular drug delivery nanocarriers of glutathione-responsive degradable block copolymers having pendant disulfide linkages, Biomacromolecules, 2013, 14(6), 2103–2111 CrossRef CAS PubMed.
- W. Yuan, Z. Lu, J. Liu, H. Wang and C. M. Li, ZnO nanowire array-templated LbL self-assembled polyelectrolyte nanotube arrays and application for charged drug delivery, Nanotechnology, 2013, 24(4), 045605 CrossRef PubMed.
- W. Yuan, Z. Lu and C. M. Li, Controllably layer-by-layer self-assembled polyelectrolytes/nanoparticle blend hollow capsules and their unique properties, J. Mater. Chem., 2011, 21(13), 5148–5155 RSC.
- U. Kedar, P. Phutane, S. Shidhaye and V. Kadam, Advances in polymeric micelles for drug delivery and tumor targeting, Nanomed. Nanotechnol. Biol. Med., 2010, 6, 714–729 CrossRef CAS PubMed.
- M. C. Jones and J. C. Leroux, Polymeric micelles–a new generation of colloidal drug carriers, Eur. J. Pharm. Biopharm., 1999, 48(2), 101–111 CrossRef CAS.
- K. Miyata, R. J. Christie and K. Kataoka, Polymeric micelles for nano-scale drug delivery, React. Funct. Polym., 2011, 71(3), 227–234 CrossRef CAS PubMed.
- V. P. Torchilin, PEG-based micelles as carriers of contrast agents for different imaging modalities, Adv. Drug Delivery Rev., 2002, 54, 235–252 CrossRef CAS.
- A. N. Lukyanov and V. P. Torchilin, Micelles from lipid derivatives of water-soluble polymers as delivery systems for poorly soluble drugs, Adv. Drug Delivery Rev., 2004, 56, 1273–1289 CrossRef CAS PubMed.
- Y.-Y. Diao, H.-Y. Li, Y.-H. Fu, M. Han, Y.-L. Hu, H.-L. Jiang, Y. Tsutsumi, Q.-C. Wei, D.-W. Chen and J.-Q. Gao, Doxorubicin-loaded PEG-PCL copolymer micelles enhance cytotoxicity and intracellular accumulation of doxorubicin in adriamycin-resistant tumor cells, Int. J. Nanomed., 2011, 6, 1955–1962 CAS.
- J.-H. Ryu, R. Roy, J. Ventura and S. Thayumanavan, Redox-Sensitive Disassembly of Amphiphilic Copolymer Based Micelles, Langmuir, 2010, 26, 7086–7092 CrossRef CAS PubMed.
- Y. Q. Yang, X. D. Guo, W. J. Lin, L. J. Zhang, C. Y. Zhang and Y. Qian, Amphiphilic copolymer brush with random pH-sensitive/hydrophobic structure: synthesis and self-assembled micelles for sustained drug delivery, Soft Matter, 2012, 8(2), 454–464 RSC.
- K. Park, J. H. Kim, Y. S. Nam, S. Lee, H. Y. Nam, K. Kim and I. C. Kwon, Effect of polymer molecular weight on the tumor targeting characteristics of self-assembled glycol chitosan nanoparticles, J. Controlled Release, 2007, 122(3), 305–314 CrossRef CAS PubMed.
- R. B. P. E. G. Greenwald, drugs: an overview, J. Controlled Release, 2001, 74, 159–171 CrossRef CAS.
- G. Prencipe, S. M. Tabakman, K. Welsher, Z. Liu, A. P. Goodwin, L. Zhang, J. Henry and H. Dai, PEG branched polymer for functionalization of nanomaterials with ultralong blood circulation, J. Am. Chem. Soc., 2009, 131, 4783–4787 CrossRef CAS PubMed.
- G. Kwon, S. Suwa, M. Yokoyama, T. Okano, Y. Sakurai and K. Kataoka, Enhanced tumor accumulation and prolonged circulation times of micelle-forming poly (ethylene oxide-aspartate) block copolymer-adriamycin conjugates, J. Controlled Release, 1994, 29, 17–23 CrossRef CAS.
- M. J. Joralemon, S. McRae and T. Emrick, PEGylated polymers for medicine: from conjugation to self-assembled systems, Chem. Commun., 2010, 46(9), 1377–1393 RSC.
- Y. Mai and A. Eisenberg, Self-assembly of block copolymers, Chem. Soc. Rev., 2012, 41(18), 5969–5985 RSC.
- E. R. Gillies and J. M. Frechet, Dendrimers and dendritic polymers in drug delivery, Drug Discovery Today, 2005, 10(1), 35–43 CrossRef CAS.
- T. Noda and Y. Morishima, Hydrophobic association of random copolymers of sodium 2-(acrylamido)-2-methylpropanesulfonate and dodecyl methacrylate in water as studied by fluorescence and dynamic light scattering, Macromolecules, 1999, 32(14), 4631–4640 CrossRef CAS.
- D. Fournier, R. Hoogenboom, H. M. Thijs, R. M. Paulus and U. S. Schubert, Tunable pH-and temperature-sensitive copolymer libraries by reversible addition-fragmentation chain transfer copolymerizations of methacrylates, Macromolecules, 2007, 40(4), 915–920 CrossRef CAS.
- X. Zhu and M. Liu, Self-assembly and morphology control of new L-glutamic acid-based amphiphilic random copolymers: Giant vesicles, vesicles, spheres, and honeycomb film, Langmuir, 2011, 27(21), 12844–12850 CrossRef CAS PubMed.
- M. A. Schott, M. Domurado, L. Leclercq, C. Barbaud and D. Domurado, Solubilization of Water-Insoluble Drugs Due to Random Amphiphilic and Degradable Poly (dimethylmalic acid) Derivatives, Biomacromolecules, 2013, 14(6), 1936–1944 CrossRef CAS PubMed.
- H. S. Kang, S. R. Yang, J. D. Kim, S. H. Han and I. S. Chang, Effects of grafted alkyl groups on aggregation behavior of amphiphilic poly (aspartic acid), Langmuir, 2001, 17(24), 7501–7506 CrossRef CAS.
- Z. Zhang, Q. Qu, J. Li and S. Zhou, The effect of the hydrophilic/hydrophobic ratio of polymeric micelles on their endocytosis pathways into cells, Macromol. Biosci., 2013, 13(6), 789–798 CrossRef CAS PubMed.
- H. Yamamoto, M. Mizusaki, K. Yoda and Y. Morishima, Fluorescence Studies of Hydrophobic Association of Random Copolymers of Sodium 2-(Acrylamido)-2-methylpropanesulfonate and N-dodecylmethacrylamide in Water, Macromolecules, 1998, 31, 3588–3594 CrossRef CAS.
- K. Y. Lee, W. H. Jo, I. C. Kwon, Y. H. Kim and S. Y. Jeong, Structural determination and interior polarity of self-aggregates prepared from deoxycholic acid-modified chitosan in water, Macromolecules, 1998, 31(2), 378–383 CrossRef CAS.
- H. Ringsdorf, J. Venzmer and F. M. Winnik, Fluorescence studies of hydrophobically modified poly (N-isopropylacrylamides), Macromolecules, 1991, 24(7), 1678–1686 CrossRef CAS.
- M. Suwa, A. Hashidzume, Y. Morishima, T. Nakato and M. Tomida, Self-association behavior of hydrophobically modified poly (aspartic acid) in water studied by fluorescence and dynamic light scattering techniques, Macromolecules, 2000, 33(21), 7884–7892 CrossRef CAS.
- N. Rapoport, Physical stimuli-responsive polymeric micelles for anti-cancer drug delivery, Prog. Polym. Sci., 2007, 32, 962–990 CrossRef CAS PubMed.
- C. J. F. Rijcken, O. Soga, W. E. Hennink and C. F. van Nostrum, Triggered destabilisation of polymeric micelles and vesicles by changing polymers polarity: An attractive tool for drug delivery, J. Controlled Release, 2007, 120, 131–148 CrossRef CAS PubMed.
- A. Mihranyan, N. Ferraz and M. Strømme, Current status and future prospects of nanotechnology in cosmetics, Prog. Mater. Sci., 2012, 57(5), 875–910 CrossRef CAS PubMed.
- G. Han, C.-C. You, B.-j. Kim, R. S. Turingan, N. S. Forbes, C. T. Martin and V. M. Rotello, Light-Regulated Release of DNA and Its Delivery to Nuclei by Means of Photolabile Gold Nanoparticles, Angew. Chem., Int. Ed., 2006, 45, 3165–3169 CrossRef CAS PubMed.
- V. Yesilyurt, R. Ramireddy and S. Thayumanavan, Photoregulated Release of Noncovalent Guests from Dendritic Amphiphilic Nanocontainers, Angew. Chem., Int. Ed., 2011, 50, 3038–3042 CrossRef CAS PubMed.
- J. Jiang, X. Tong, D. Morris and Y. Zhao, Toward Photocontrolled Release Using Light-Dissociable Block Copolymer Micelles, Macromolecules, 2006, 39, 4633–4640 CrossRef CAS.
- D. Schmaljohann, Thermo- and pH-responsive polymers in drug delivery, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
- Y. He, Y. Zhang, Y. Xiao and M. Lang, Dual-response nanocarrier based on graft copolymers with hydrazone bond linkages for improved drug delivery, Colloids Surf., B, 2010, 80, 145–154 CrossRef CAS PubMed.
- G. Li, S. Song, L. Guo and S. Ma, Self-Assembly of Thermo- and pH-Responsive Poly(acrylic acid)-b-poly(N isopropylacrylamide) Micelles for Drug Delivery, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5028–5035 CrossRef CAS.
- W. Yuan, Z. Lu and C. M. Li, Charged drug delivery by ultrafast exponentially grown weak polyelectrolyte multilayers: amphoteric properties, ultrahigh loading capacity and pH-responsiveness, J. Mater. Chem., 2012, 22(18), 9351–9357 RSC.
- M. F. Francis, M. Piredda and F. M. Winnik, Solubilization of poorly water soluble drugs in micelles of hydrophobically modified hydroxypropylcellulose copolymers, J. Controlled Release, 2003, 93(1), 59–68 CrossRef CAS PubMed.
- J. O’Leary and F. M. Muggia, Camptothecins: a review of their development and schedules of administration, Eur. J. Cancer, 1998, 34, 1500–1508 CrossRef.
- J. Dey and I. M. Warner, Excited state tautomerization of camtothecin in aqueous solution, J. Photochem. Photobiol., A, 1996, 101, 21–27 CrossRef CAS.
- P. Opanasopit, T. Ngawhirunpat, A. Chaidedgumjorn, T. Rojanarata, A. Apirakaramwong, S. Phongying, C. Choochottiros and S. Chirachanchai, Incorporation of camptothecin into N-phthaloyl chitosan-g-mPEG self-assembly micellar system, Eur. J. Pharm. Biopharm., 2006, 64, 269–276 CrossRef CAS PubMed.
- Z. Mi and T. G. Burke, Differential Interactions of Camptothecin Lactone and Carboxylate Forms with Human Blood Components, Biochemistry, 1994, 33, 10325–10336 CrossRef CAS.
- M. B. Milovanovic, M. Avramovic, L. Katsikas and I. G. Popovic, Simplification of the synthesis of the reversible addition–fragmentation chain transfer agent 2-(2-cyanopropyl)-dithiobenzoate, J. Serb. Chem. Soc., 2010, 75(12), 1711–1719 CrossRef CAS.
- S. Perrier, C. Barner-Kowollik, J. F. Quinn, P. Vana and T. P. Davis, Origin of inhibition effects in the reversible addition fragmentation chain transfer (RAFT) polymerization of methyl acrylate, Macromolecules, 2002, 35(22), 8300–8306 CrossRef CAS.
- P. Dutta and J. Dey, Drug solubilization by amino acid based polymeric nanoparticles: Characterization and biocompatibility studies, Int. J. Pharm., 2011, 421, 353–363 CrossRef CAS PubMed.
- M. E. H. El-Sayed, A. S. Hoffman and P. S. Stayton, Rational design of composition and activity correlations for pH-sensitive and glutathione-reactive polymer therapeutics, J. Controlled Release, 2005, 101, 47–58 CrossRef CAS PubMed.
- A. Sulistio, J. Lowenthal, A. Blencowe, M. N. Bongiovanni, L. Ong, S. L. Gras, X. Zhang and G. G. Qiao, Folic Acid Conjugated Amino Acid-Based Star Polymers for Active Targeting of Cancer Cells, Biomacromolecules, 2011, 12, 3469–3477 CrossRef CAS PubMed.
- K. Kalyanasundaram and J. K. Thomas, Environmental effects on vibronic band intensities in pyrene monomer fluorescence and their application in studies of micellar systems, J. Am. Chem. Soc., 1977, 99, 2039–2044 CrossRef CAS.
- P. Dutta, S. Shrivastava and J. Dey, Amphiphilic polymer nanoparticles: characterization and assessment as new drug carriers, Macromol. Biosci., 2009, 9(11), 1116–1126 CrossRef CAS PubMed.
- K. E. Schmalenberg, L. Frauchiger, L. Nikkhouy-Albers and K. E. Uhrich, Cytotoxicity of a Unimolecular Polymeric Micelle and Its Degradation Products, Biomacromolecules, 2001, 2, 851–855 CrossRef CAS.
- G. Storm, S. Belliot, T. Daemen and D. Lasic, Surface modification of nanoparticles to oppose uptake by the mononuclear phagocyte system, Adv. Drug Delivery Rev., 1995, 17, 31–48 CrossRef CAS.
- S. Ghosh, K. Irvin and S. Thayumanavan, Tunable Disassembly of Micelles Using a Redox Trigger, Langmuir, 2007, 23, 7916–7919 CrossRef CAS PubMed.
- Y. Huang, H. Yu, L. Guo and Q. Huang, Structure and Self-Assembly Properties of a New Chitosan-Based Amphiphile, J. Phys. Chem. B, 2010, 114, 7719–7726 CrossRef CAS PubMed.
- T. Akagi, P. Piyapakorn and M. Akashi, Formation of unimer nanoparticles by controlling the self-association of hydrophobically modified poly (amino acid)s, Langmuir, 2012, 28(11), 5249–5256 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Plenum Press, New York, 1983, p. 132 Search PubMed.
- P. Debye, Polar Molecules, Dover, New York, 1929 Search PubMed.
- S. Roy, A. Mohanty and J. Dey, Microviscosity of bilayer membranes of some N-acyl amino acid surfactants determined by fluorescence probe method, Chem. Phys. Lett., 2005, 414, 23–27 CrossRef CAS PubMed.
- R. B. Greenwald, Y. H. Choe, J. McGuire and C. D. Conover, Effective drug delivery by PEGylated drug conjugates, Adv. Drug Delivery Rev., 2003, 55, 217–250 CrossRef CAS.
- D. Yu, P. Peng, S. S. Dharap, Y. Wang, M. Mehlig, P. Chandna, H. Zhao, D. Filpula, K. Yang, V. Borowski, G. Borchard, Z. Zhang and T. Minko, Antitumor activity of poly (ethylene glycol)–camptothecin conjugate: The inhibition of tumor growth in vivo, J. Controlled Release, 2005, 110, 90–102 CrossRef CAS PubMed.
- J. Dey and I. M. Warner, Spectroscopic and photophysical studies of the anticancer drug: Camptothecin, J. Lumin., 1997, 71, 105–114 CrossRef CAS.
- S. V. Aathimanikandan, E. N. Savariar and S. Thayumanavan, Temperature-sensitive dendritic micelles, J. Am. Chem. Soc., 2005, 127(42), 14922–14929 CrossRef CAS PubMed.
- P. Laskar, S. Samanta, S. K. Ghosh and J. Dey, In vitro Evaluation of pH-Sensitive Cholesterol-Containing Stable Polymeric Micelles for Delivery of Camptothecin, J. Colloid Interface Sci., 2014, 430, 305–314 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the synthesis and chemical identification of the polymers by 1H-NMR spectroscopy, surface tension plots, fluorescence spectra, lifetime data, and hydrodynamic size distributions. See DOI: 10.1039/c4ra11479e |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.