
One-pot synthesis of magnetic molecularly imprinted microspheres by RAFT precipitation polymerization for the fast and selective removal of 17β-estradiol†
 *a
*a
Abstract
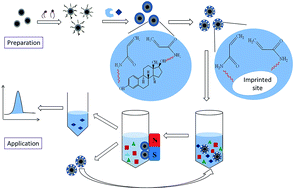
A facile strategy was developed to prepare magnetic molecularly imprinted microspheres (MMIMs) for the selective recognition and effective removal of 17-beta-estradiol (17β-E2) by reversible addition-fragmentation chain transfer precipitation polymerization. One-pot synthesis was employed, which could simplify the imprinting process and shorten the experimental period. The resultant MMIMs displayed fast kinetics and high binding capacity, and the adsorption processes followed Langmuir–Freundlich isotherm and pseudo-second-order kinetic models. Excellent recognition selectivity toward 17β-E2 was attained over other phenolic estrogens such as 17-alpha-E2, estriol and estrone. The magnetic property of MMIMs provided fast and simple separation, and the recycling process for magnetic solid phase extraction (MSPE) was sustainable at least five times without obvious efficiency decrease. Furthermore, the MMIMs-MSPE presented satisfactory recoveries within 71.7–108.3% with the precisions of 1.1–6.0% for spiked 17β-E2 in water, soil and food samples. The developed MMIMs-based method proved to be a convenient and practical way in sample pretreatment and targeted pollutants removal.
 Please wait while we load your content...
Please wait while we load your content...

