
Novel carbazole-based two-photon photosensitizer for efficient DNA photocleavage in anaerobic condition using near-infrared light†
Yong-Chao Zhengad,
Mei-Ling Zheng*a,
Ke Libd,
Shu Chena,
Zhen-Sheng Zhaoa,
Xue-Song Wangb and
Xuan-Ming Duan*ac
aLaboratory of Organic NanoPhotonics and Key Laboratory of Functional Crystals and Laser Technology, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, No. 29 Zhongguancun East Road, Beijing 100190, P. R. China. E-mail: zhengmeiling@mail.ipc.ac.cn; xmduan@mail.ipc.ac.cn; Fax: +86-10-82543597; Tel: +86-10-82543596
bKey Laboratory of Photochemical Conversion and Optoelectronic Materials, Technical Institute of Physics and Chemistry, Chinese Academy of Sciences, No. 29 Zhongguancun East Road, Beijing 100190, P. R. China
cChongqing Institute of Green and Intelligent Technology, Chinese Academy of Sciences, No. 266 Fangzheng Ave, Shuitu Technology Development Zone, Beibei District, Chongqing 400714, P. R. China
dUniversity of Chinese Academy of Sciences, No. 29 Zhongguancun East Road, Beijing 100190, P. R. China
First published on 19th November 2014
Abstract
Two novel carbazole derivatives, BMEPC and BMEMC, were designed, synthesized and first reported as two-photon photosensitizers for DNA photodamage, which showed efficient DNA photocleavage ability under near-infrared light exposure via a two-photon process in anaerobic condition.
Photodynamic therapy (PDT), as one of the minimally invasive radiation therapy technologies for malignant tumors, has attracted great interest due to the potential lower toxicity and higher targeting by using a non-toxic photosensitizer.1 A potentially important mechanism of PDT is that photosensitizers can cause DNA photodamage in cancer cells when exposed to light with an appropriate wavelength.2 Photosensitizers such as metal complexes, porphyrins, anthraquinones, fullerenes etc. have been confirmed to possess DNA photocleavage activity through type I or type II mechanisms.3 In contrast to type II mechanism that greatly depends on O2 to generate reactive oxygen species, type I mechanism can still work at low oxygen concentrations through electron transfer or hydrogen abstraction processes, which is of great potential for extending PDT applications into hypoxic cellular areas of solid tumor tissue.4
However, most photosensitizers suffer the disadvantage of weak absorption in the phototherapeutic window of 600–900 nm, limiting their further application in PDT.5 Thus, photosensitizers with a two-photon absorption (TPA) property are very attractive since the photoactivation achieved by two-photon excitation using near-infrared (NIR) light is capable of achieving deep tissue penetration.6 Moreover, the nonlinear process of TPA of the laser intensity restricts the absorption to the focus of a laser beam, which is beneficial to highly selective targeting damage.7 Nevertheless, there have been relatively few studies on photosensitizers for PDT or photosensitized DNA damage operating through the TPA process because conventional photosensitizers usually have a very low TPA cross section (δTPA), generally of the order of 1–100 GM (1 GM = 10−50 cm4 s per photon per molecule).8 Thus, DNA photocleavers with large δTPA are needed for developing agents for two-photon excited PDT.
As known nonlinear materials, carbazole derivatives have been widely used in micro/nanofabrication, optical power limiting and bioanalytical science.9 In previous work, we have found carbazole-based cyanines can be employed as two-photon excited fluorescent (TPEF) probes for DNA and cell imaging considering their high binding affinity to DNA, large TPA cross section, and good water solubility.10 Considering the DNA photodamage ability of some carbazole derivatives on UV-light exposure, we expected that a carbazole-based molecule with large δTPA and potential interaction with DNA would photosensitize DNA damage under NIR light.11 Herein, two novel carbazole derivatives, 3,6-bis[2-(1-methylpyridinium)ethynyl]-9-pentylcarbazole diiodide (BMEPC) and 3,6-bis[2-(1-methylpyridinium)ethynyl]-9-methylcarbazole diiodide (BMEMC), were designed and synthesized (Scheme 1). The C2v symmetric A–π–D–π–A structure, strong intramolecular change transfer and planar molecular structure with positive charge contribute to large δTPA, low fluorescence quantum yield and high binding affinity towards DNA by intercalation mode.11a,12 They show efficient DNA photocleavage ability excited not only by visible light but also by 800 nm NIR light no matter in aerobic or anaerobic condition via type I mechanism.
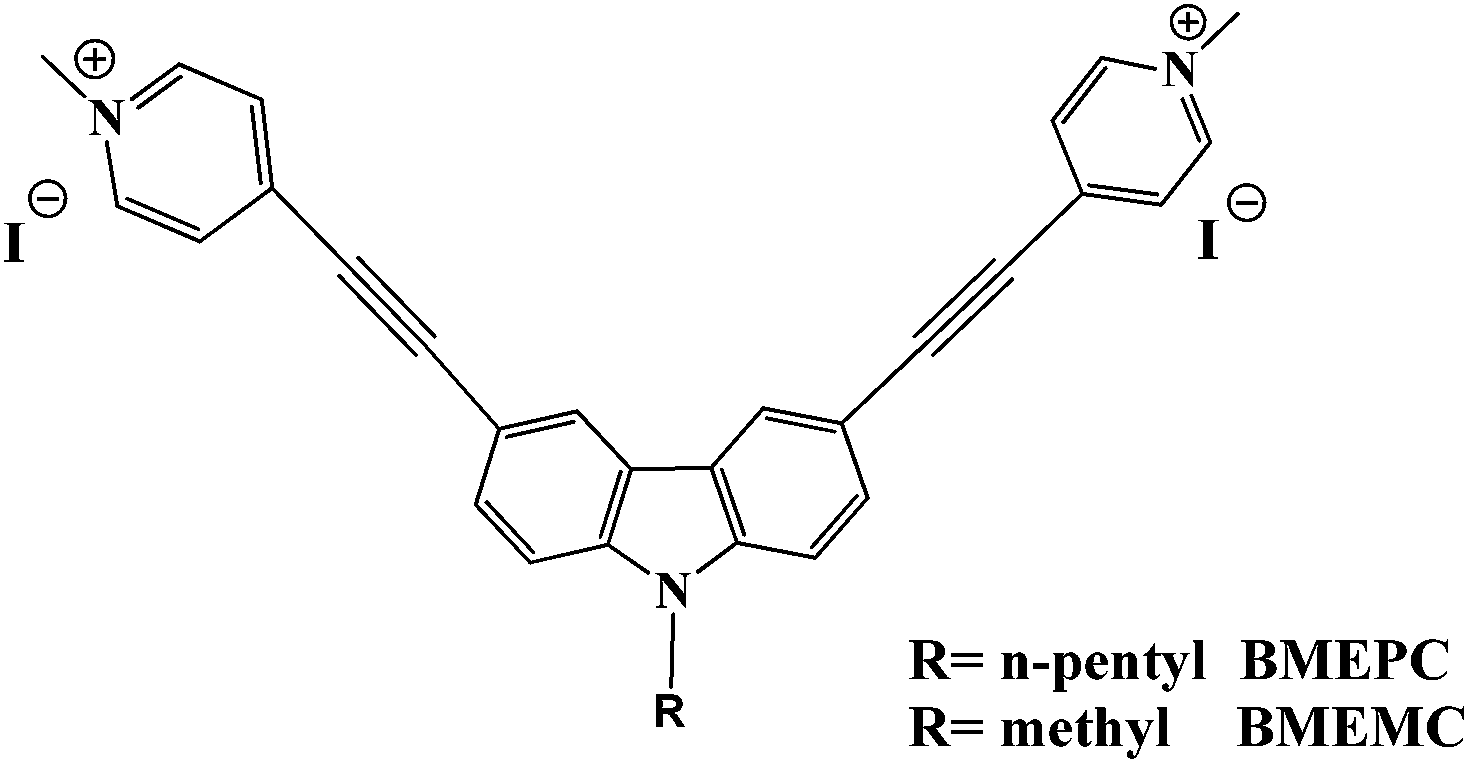 | ||
| Scheme 1 The chemical structures of BMEPC and BMEMC. | ||
The synthesis route of these carbazole-based compounds is presented in Scheme S1.† Starting from 3,6-dibromo-9H-carbazole, BMEPC and BMEMC were synthesized via the Sonogashira reaction to afford bis-ethynylpyridine carbazole according to our previous work.13 Subsequent methylation was performed to give the desired product.
The ionic groups introduced in the molecule by salification reaction are better electron acceptor groups.
The photophysical properties of BMEPC and BMEMC have been investigated and the data are summarized in Table 1. The normalized absorption and fluorescence spectra of BMEPC in dimethyl sulfoxide (DMSO) and Tris–EDTA (TE) buffer are shown in Fig. 1a. The absorption band at 418 nm is attributed to the electronic transition from the ground state to the intramolecular charge transfer (ICT) state, and the peak at 331 nm is assigned to the typical π–π* transition corresponding to the locally excited state. Besides these two peaks, there is a weak shoulder peak at 380 nm, which is attributed to the coupling of two ICT branches. The emission peaks are located at ∼590 nm with excitation wavelength of 415 nm. The fluorescence quantum yields of BMEPC and BMEMC are too low to be calculated accurately either in DMSO or buffer solution. This is most likely induced by the ICT state which mainly deactivates through a nonradiative decay, resulting in more efficient transition from singlet state to triplet state.14
| Solvent | λabsa (nm) | 104 εb (M−1 cm−1) | λemc (nm) | Φd | δe (GM) | |
|---|---|---|---|---|---|---|
| a The wavelength of absorption maximum.b The extinction coefficient.c The wavelength of one-photon emission maximum.d Fluorescence quantum yield.e TPA cross section (1 GM = 10−50 cm4 s per photon per molecule).f TPA cross section (1 GM = 10−50 cm4 s per photon per molecule) at 760 nm.g TPA cross section (1 GM = 10−50 cm4 s per photon per molecule) at 800 nm. | ||||||
| BMEPC | DMSO | 419 | 4.69 | 591 | <0.001 | 522f/401g |
| TE buffer | 418 | 4.55 | 584 | <0.001 | ||
| BMEMC | DMSO | 418 | 4.59 | 593 | <0.001 | 492f/352g |
| TE buffer | 417 | 4.58 | 583 | <0.001 | ||
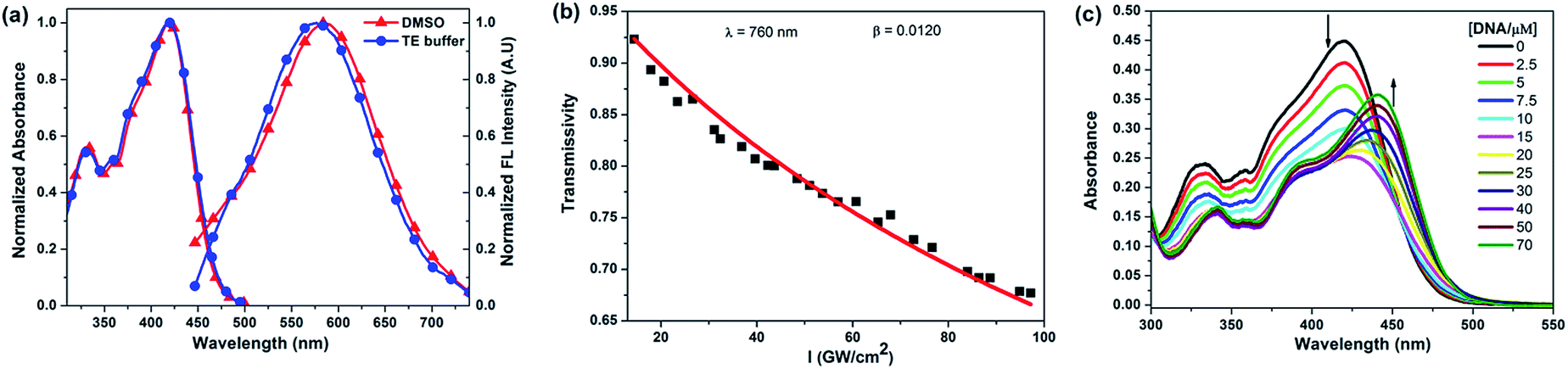 | ||
| Fig. 1 (a) Normalized absorption and one-photon-induced fluorescence spectra of BMEPC in DMSO and TE buffer. (b) The plot of dependence of transmissivity on light intensity for BMEPC at 760 nm. Squares denote the experimental values of transmissivity; solid line denotes the theoretical fitting line. (c) The absorption spectra of BMEPC (10 μM) with the addition of CT-DNA (0–70 μM) in TE buffer. | ||
Both BMEPC and BMEMC exhibit weak TPEF signals upon excitation with 800 nm femtosecond (fs) laser pulses in DMSO. Since the two-photon-induced fluorescence method is not suitable for the δTPA measurement of the two compounds due to their extremely low fluorescence quantum yields, δTPA were determined by a nonlinear transmission measurement technique using an amplified Ti:sapphire ultrafast laser system (Spitfire ACE, Spectra-Physics) at wavelengths from 750 to 810 nm.15 The relationships between transmissivity and light intensity for BMEPC and BMEMC at 760 nm and 800 nm are shown in Fig. 1b and S2, ESI.† Both of them showed a maximum δTPA at 760 nm (522 GM for BMEPC and 492 GM for BMEMC) which corresponded to the shoulder peak in one-photon absorption spectra at 380 nm as the coupling of a second ICT state. The good TPA properties imply potential applications of BMEPC and BMEMC for two-photon photosensitization.
The interaction of BMEPC and BMEMC with DNA was investigated by absorption titration. The absorption spectra of BMEPC and BMEMC upon addition of calf thymus DNA (CT-DNA) at different concentrations in TE buffer are very similar (Fig. 1c and S3, ESI†). An obvious decrease of the absorption intensity (42% and 36%, respectively) accompanied with negligible bathochromic shift for low concentration of DNA and then hyperchromism with a bathochromic shift (∼20 nm) at high concentration were observed. The spectra changes as a function of the concentration of DNA indicate that the interaction of BMEPC and BMEMC with DNA is a complex process, including at least two binding modes. The compounds may aggregate on the surface of the CT-DNA helix at low DNA concentration by electrostatic interactions and then intercalate into CT-DNA base pairs at high DNA/compound ratios leading to the increased absorbance and large red-shift of λmax, which is basically in line with the result as reported in the literature.16
The binding capabilities of BMEPC and BMEMC towards CT-DNA were investigated by fluorescence titration (Fig. S4, ESI†). The compounds showed obvious fluorescence enhancement upon the addition of CT-DNA, which is attributed to a reduction of the nonradiative decay caused by the restricted intramolecular rotation after interacting with DNA. The binding constants (Kb) of BMEPC and BMEMC with CT-DNA estimated by nonlinear curve fitting analysis are 2.9 × 105 M−1 and 3.3 × 105 M−1, respectively, which are comparable to those of some other DNA-binding molecules mentioned in the literature.17 This result indicates that the compounds possess high binding affinity to DNA due to their symmetric bis-cationic and planar structures, which implies their potential as DNA photocleavers.
BMEPC and BMEMC were applied to the photocleavage of supercoiled pBR322 DNA upon light irradiation in air-saturated buffer. As shown in Fig. 2, the plasmid is in the supercoiled form (Form I) with a small amount of nicked circular form (Form II) in the absence of compounds (lanes 1 and 2). DNA was not cleaved in the presence of the compounds (20 μM) in the dark (lanes 4 and 6). Under light irradiation (λ > 400 nm, 25 min), BMEPC and BMEMC can lead to single-strand DNA cleavage, as evidenced by the transformation from Form I to Form II (lanes 3 and 5). The results show the light radiation is necessary for DNA cleavage by BMEPC and BMEMC.
 | ||
| Fig. 2 Agarose gel electrophoresis patterns of photocleaved supercoiled pBR322 DNA (31 μM in base pair) using BMEPC and BMEMC (20 μM) upon visible-light irradiation (>400 nm) for 25 min in air-saturated Tris/CH3COOH/EDTA buffer (pH = 7.4). Lane 1, DNA alone (in dark); lane 2, DNA + irradiation; lane 3, DNA + BMEPC + irradiation; lane 4, DNA + BMEPC (in dark); lane 5, DNA + BMEMC + irradiation; lane 6, DNA + BMEMC (in dark). Forms I and II denote supercoiled circular and nicked circular forms, respectively. | ||
We further examined the DNA photocleavage activity of BMEPC through a two-photon absorption process using an 800 nm laser pulse with a pulse width of 120 fs and a repetition rate of 1 kHz. As shown in Fig. 3a, supercoiled pBR322 DNA in different concentrations of BMEPC was irradiated for 35 min under a defocused laser beam with an average power of 0.3 W cm−2. Control experiment proves that the presence of BMEPC is necessary for DNA cleavage (Fig. 3b). Less DNA cleavage was observed for 10 μM BMEPC upon irradiation, while DNA converted completely from supercoiled circular form to nicked circular form at a concentration of 30 μM (lanes 2 and 4). These results indicate that the high DNA photocleavage activities of BMEPC and BMEMC can be induced by not only one-photon absorption process, but also TPA process, which enable them to be excited with a NIR light source.
 | ||
| Fig. 3 (a) Experimental setup for two-photon DNA photocleavage. (b) Agarose gel electrophoresis patterns of photocleaved supercoiled pBR322 DNA (31 μM in base pair) by BMEPC upon 800 nm femtosecond (fs) laser (0.3 W cm−2) irradiation for 35 min in air-saturated Tris/CH3COOH/EDTA buffer (pH = 7.4). Lane 1, DNA alone; lane 2, DNA + BMEPC (10 μM); lane 3, DNA + BMEPC (20 μM); lane 4, DNA + BMEPC (30 μM); lane 5, DNA + BMEPC (40 μM); lane 6, DNA + BMEPC (50 μM). Forms I and II denote supercoiled circular and nicked circular forms, respectively. | ||
To investigate the possible mechanism of the photosensitized DNA damage by BMEPC, a control experiment performed in N2 atmosphere showed that there was no significant difference for the DNA photocleavage results between aerobic and anaerobic conditions (Fig. S5, ESI†). The results suggest that the photocleavage mainly results from type I mechanism as oxygen is not an essential cofactor for DNA photocleavage which is also in agreement with the very low efficiency in singlet oxygen production of BMEPC (data not shown).18 Furthermore, negligible inhibiting effect for DNA cleavage was observed in the presence of superoxide dismutase and mannitol, indicating that superoxide anions (O2−˙) and hydroxyl radicals (˙OH) were not involved (Fig. S5, ESI†). Electron transfer from guanine to the photoexcited BMEPC can be excluded on account of the relatively short excited state lifetime of BMEPC (<ns level) and it is also impossible in terms of energy according to the estimated Gibbs free energy (+0.15 eV) of the process (Fig. S6, ESI†).19 A possible explanation for the photocleavage activity of BMEPC is the reactive intermediates generated by photochemical effects. The planar carbazole molecule with pyridinium cations as strong acceptor group can form N-centered radical cations through electron transfer.20 As shown in Fig. 4a, the EPR signals of BMEPC using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as spin trapping agent can be assigned to aminyl radicals, which is in good accordance with results reported in the literature.20b,21 Similar signals were also obtained in PBS solution in the presence of pBR322 DNA (Fig. 4b). The amine radical cations generated by photoirradiation may abstract hydrogen from the adjacent deoxyribose, leading to the DNA cleavage.21,22 In addition, the inhibiting effect for DNA cleavage in the presence of NaN3 was likely caused by the quenching of the radical species, which is reasonable in that NaN3 is a singlet oxygen scavenger but not strictly specific.23
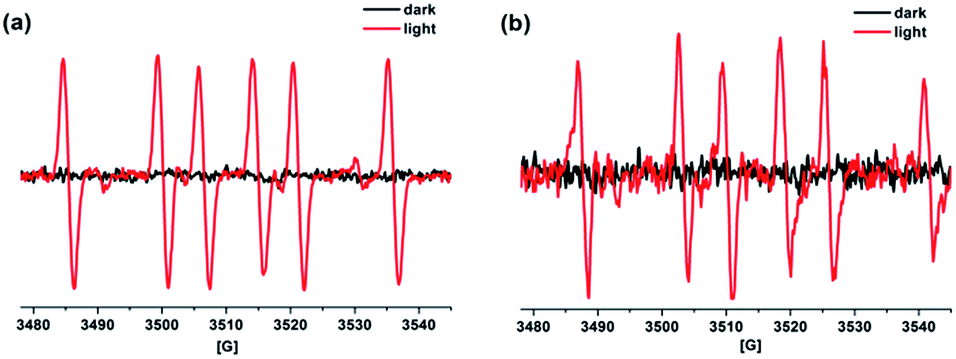 | ||
| Fig. 4 EPR spectra of DMPO spin adducts. (a) N2-saturated DMSO solutions of 1 mM BMEPC with mercury lamp; the splitting parameters are g = 2.0046, aNα = 14.5 G and aHβ = 21.1 G. (b) N2-saturated PBS (pH 7.4) solutions of 50 μM BMEPC and pBR322 DNA (31 μM in base pair) with mercury lamp; the splitting parameters are g = 2.0063, aNα = 15.6 G and aHβ = 22.3 G. Dark control means the sample without light irradiation. | ||
In summary, we have successfully designed and synthesized two novel carbazole derivatives, BMEPC and BMEMC, as photosensitizers for DNA photocleavage. The molecular structure characteristics contribute to the large TPA cross section and the high binding affinity towards DNA. DNA photocleavage can be achieved efficiently in the presence of BMEPC and BMEMC excited not only by visible light but also 800 nm NIR light through a TPA process. The experimental evidence supports the fact that BMEPC and BMEMC photocleave DNA mainly via hydrogen abstraction by N-centered radicals (type I mechanism), contributing to the DNA photocleavage ability in anaerobic conditions. Such carbazole-based photocleavers are valuable for the development of new two-photon excited PDT agents. Further studies are underway to investigate the detailed mechanism, cytotoxicity and PDT experiment in vivo.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (grant no. 61475164 and 61205194), CAS-JSPS Joint Research Project (GJHZ1411), and the National Basic Research Program of China (2010CB934103).Notes and references
- (a) D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380 CrossRef CAS; (b) A. P. Castano, P. Mroz and M. R. Hamblin, Nat. Rev. Cancer, 2006, 6, 535 CrossRef CAS.
- (a) K. Kawai, Y. Osakada, M. Fujitsuka and T. Majima, J. Phys. Chem. B, 2007, 111, 2322 CrossRef CAS PubMed; (b) A. Ikeda, Y. Doi, M. Hashizume, J.-i. Kikuchi and T. Konishi, J. Am. Chem. Soc., 2007, 129, 4140 CrossRef CAS PubMed.
- (a) Y. Sun, L. E. Joyce, N. M. Dickson and C. Turro, Chem. Commun., 2010, 46, 2426 RSC; (b) C. Zhou, Q. Liu, W. Xu, C. Wang and X. Fang, Chem. Commun., 2011, 47, 2982 RSC; (c) V. Singh, P. C. Mondal, A. Kumar, Y. L. Jeyachandran, S. K. Awasthi, R. D. Gupta and M. Zharnikov, Chem. Commun., 2014, 50, 11484 RSC; (d) T. Da Ros, G. Spalluto, A. S. Boutorine, R. V. Bensasson and M. Prato, Curr. Pharm. Des., 2001, 7, 1781 CrossRef CAS; (e) N. Paillous and P. Vicendo, J. Photochem. Photobiol., B, 1993, 20, 203 CrossRef CAS.
- (a) Q.-X. Zhou, W.-H. Lei, C. Li, Y.-J. Hou, X.-S. Wang and B.-W. Zhang, New J. Chem., 2010, 34, 137 RSC; (b) A. S. Karwa, A. R. Poreddy, B. Asmelash, T.-S. Lin, R. B. Dorshow and R. Rajagopalan, ACS Med. Chem. Lett., 2011, 2, 828 CrossRef CAS PubMed.
- (a) J. F. Lovell, T. W. B. Liu, J. Chen and G. Zheng, Chem. Rev., 2010, 110, 2839 CrossRef CAS PubMed; (b) M. R. Detty, S. L. Gibson and S. J. Wagner, J. Med. Chem., 2004, 47, 3897 CrossRef CAS PubMed.
- (a) J. D. Bhawalkar, N. D. Kumar, C. F. Zhao and P. N. Prasad, J. Clin. Laser Med. Surg., 1997, 15, 201 CAS; (b) K. Ogawa and Y. Kobuke, Anti-Cancer Agents Med. Chem., 2008, 8, 269 CrossRef CAS.
- F. Helmchen and W. Denk, Nat. Methods, 2005, 2, 932 CrossRef CAS PubMed.
- (a) J. R. Starkey, A. K. Rebane, M. A. Drobizhev, F. Meng, A. Gong, A. Elliott, K. McInnerney and C. W. Spangler, Clin. Cancer Res., 2008, 14, 6564 CrossRef CAS PubMed; (b) A. Karotki, M. Khurana, J. R. Lepock and B. C. Wilson, Photochem. Photobiol., 2006, 82, 443 CrossRef CAS PubMed; (c) C. Xu, R. M. Williams, W. Zipfel and W. W. Webb, Bioimaging, 1996, 4, 198 CAS.
- (a) M. D. Cahalan, I. Parker, S. H. Wei and M. J. Miller, Nat. Rev. Immunol., 2002, 2, 872 CrossRef CAS PubMed; (b) W.-E. Lu, X.-Z. Dong, W.-Q. Chen, Z.-S. Zhao and X.-M. Duan, J. Mater. Chem., 2011, 21, 5650 RSC; (c) C. W. Spangler, J. Mater. Chem., 1999, 9, 2013 RSC.
- (a) M.-L. Zheng, K. Fujita, W.-Q. Chen, N. I. Smith, X.-M. Duan and S. Kawata, ChemBioChem, 2011, 12, 52 CrossRef CAS PubMed; (b) Y.-C. Zheng, M.-L. Zheng, S. Chen, Z.-S. Zhao and X.-M. Duan, J. Mater. Chem. B, 2014, 2, 2301 RSC.
- (a) W. Sajewicz and A. Dlugosz, J. Appl. Toxicol., 2000, 20, 305 CrossRef CAS; (b) U. Jacquemard, S. Routier, A. Tatibouet, J. Kluza, W. Laine, C. Bal, C. Bailly and J.-Y. Merour, Org. Biomol. Chem., 2004, 2, 1476 RSC.
- (a) J. Gu, W. Yulan, W.-Q. Chen, X.-Z. Dong, X.-M. Duan and S. Kawata, New J. Chem., 2007, 31, 63 RSC; (b) H. Taima, A. Okubo, N. Yoshioka and H. Inoue, Chem.–Eur. J., 2006, 12, 6331 CrossRef CAS PubMed.
- J.-F. Xing, W.-Q. Chen, J. Gu, X.-Z. Dong, N. Takeyasu, T. Tanaka, X.-M. Duan and S. Kawata, J. Mater. Chem., 2007, 17, 1433 RSC.
- S. K. Lower and M. A. El-Sayed, Chem. Rev., 1966, 66, 199 CrossRef CAS.
- C. Xu and W. W. Webb, J. Opt. Soc. Am. B, 1996, 13, 481 CrossRef CAS.
- (a) Z. Li, S. Sun, Z. Yang, S. Zhang, H. Zhang, M. Hu, J. Cao, J. Wang, F. Liu, F. Song, J. Fan and X. Peng, Biomaterials, 2013, 34, 6473 CrossRef CAS PubMed; (b) X. Jiang, L. Shang, Z. Wang and S. Dong, Biophys. Chem., 2005, 118, 42 CrossRef CAS PubMed.
- J.-Y. Pang, Y. Qin, W.-H. Chen, G.-A. Luo and Z.-H. Jiang, Bioorg. Med. Chem., 2005, 13, 5835 CrossRef CAS PubMed.
- (a) A. J. Blacker, J. Jazwinski, J.-M. Lehn and F. X. Wilhelm, J. Chem. Soc., Chem. Commun., 1986, 1035 RSC; (b) D. Schulte-Frohlinde, M. G. Simic and H. Görner, Photochem. Photobiol., 1990, 52, 1137 CrossRef CAS PubMed.
- (a) K. Hirakawa, K. Ota, J. Hirayama, S. Oikawa and S. Kawanishi, Chem. Res. Toxicol., 2014, 27, 649 CrossRef CAS PubMed; (b) A.-R. Hwang, W.-S. Han, K.-R. Wee, H. Y. Kim, D. W. Cho, B. K. Min, S. W. Nam, C. Pac and S. O. Kang, J. Phys. Chem. C, 2011, 116, 1973 CrossRef; (c) G. G. Aloisi, A. Barbafina, M. Canton, F. Dall'Acqua, F. Elisei, L. Facciolo, L. Latterini and G. Viola, Photochem. Photobiol., 2004, 79, 248 CrossRef CAS PubMed.
- (a) O. Brede, A. Maroz, R. Hermann and S. Naumov, J. Phys. Chem. A, 2005, 109, 8081 CrossRef CAS PubMed; (b) R. Rajagopalan, T.-S. Lin, A. S. Karwa, A. R. Poreddy, B. Asmelash and R. B. Dorshow, ACS Med. Chem. Lett., 2012, 3, 284 CrossRef CAS PubMed.
- T.-S. Lin, R. Rajagopalan, Y. Shen, S. Park, A. R. Poreddy, B. Asmelash, A. S. Karwa and J.-S. A. Taylor, J. Phys. Chem. A, 2013, 117, 5454 CrossRef CAS PubMed.
- (a) W. K. Pogozelski and T. D. Tullius, Chem. Rev., 1998, 98, 1089 CrossRef CAS PubMed; (b) B. Armitage, Chem. Rev., 1998, 98, 1171 CrossRef CAS PubMed.
- G. G. Aloisi, M. Amelia, A. Barbafina, L. Latterini, F. Elisei, F. Dall'Acqua, D. Vedaldi, A. Faccio and G. Viola, Photochem. Photobiol., 2007, 83, 664 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S6, experimental details, compound characterization, fluorescence titration, electrochemical properties. See DOI: 10.1039/c4ra11133h |
| This journal is © The Royal Society of Chemistry 2015 |