
MoO2Cl2(DMF)2 catalyzed microwave assisted reductive cyclisation of nitroaromatics into dibenzodiazepines†
Abstract
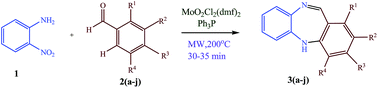
The paper describes a gentle and highly efficient protocol for the synthesis of dibenzodiazepines by reductive cyclisation of nitroaniline to dibenzodiazepines under microwave irradiation catalyzed by MoO2Cl2(DMF)2 involving Ph3P as reducing agent. This synergic approach results in the transformation of nitroaromatics into dibenzodiazepines in high yield and is applicable to the construction of a wide variety of dibenzo(di/ox)azepines and other structurally related heterocycles.
 Please wait while we load your content...
Please wait while we load your content...

