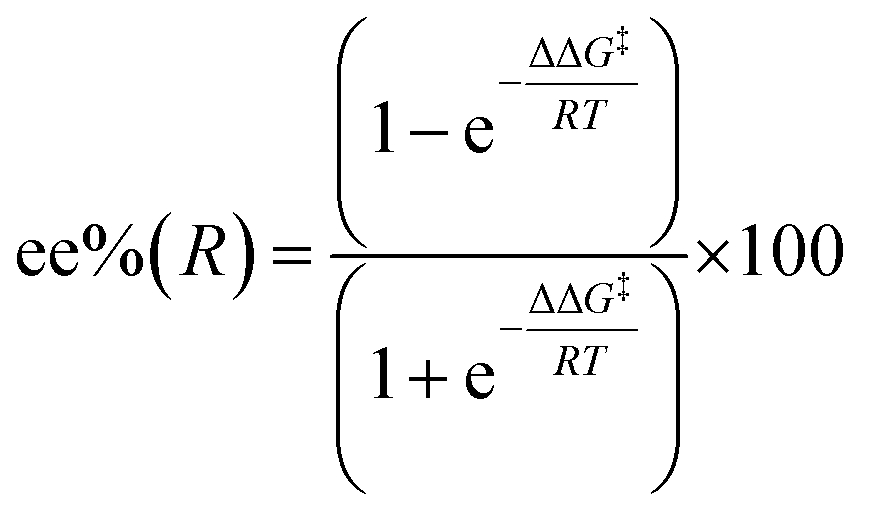DOI:
10.1039/C4RA13012J
(Paper)
RSC Adv., 2015,
5, 5250-5255
Computational modelling of the enantioselectivity in the asymmetric 1,4-addition reaction catalyzed by a Rh complex of a S-chiral disulfoxide†
Received
23rd October 2014
, Accepted 4th December 2014
First published on 4th December 2014
Abstract
Computational chemistry is a powerful tool for understanding chiral catalysis and aiding future catalyst design. Here we present a DFT (PCM/PBE0/DGDZVP) modelling of the enantioselective step in the 1,4-addition of phenylboronic acid to 5 Michael acceptors catalyzed by Rh ligated with a disulfoxide ligand whose only source of chirality is the sulfur atoms. For all substrates, the predicted absolute configuration was in agreement with the experiment. Using the dispersion-interaction-corrected SMD/M06 method improved the quantitative agreement of predicted and experimental %ee values (within 0.2–0.8 kcal mol−1). The high steric pressure exerted by the bulky tert-butyl groups is the primary determinant of the enantioselectivity.
Introduction
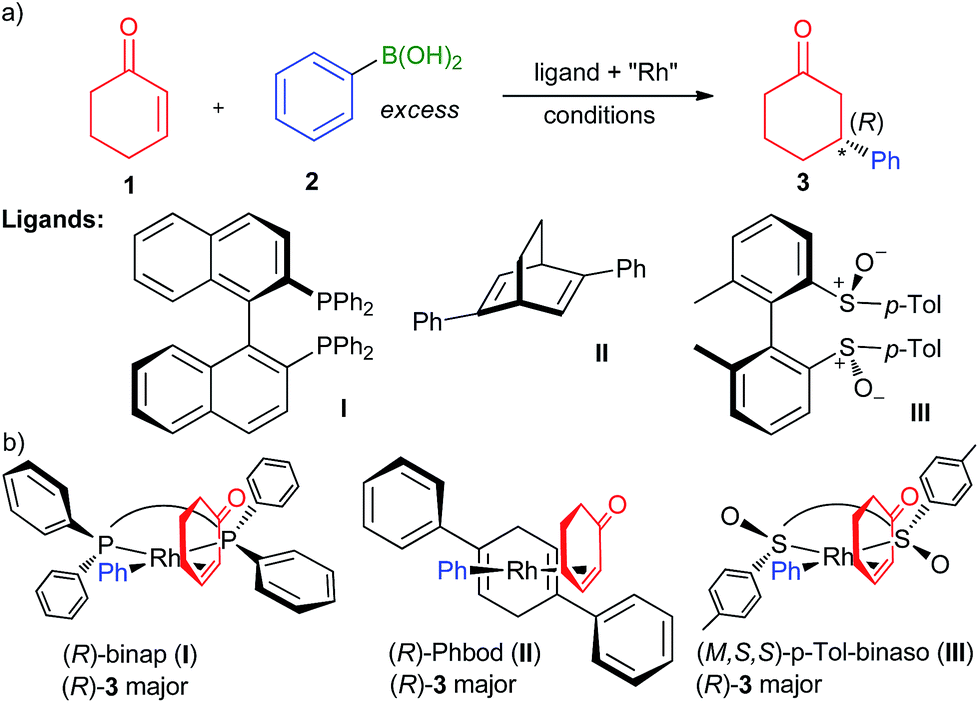
The discovery of chiral ligands for transition metal-catalyzed asymmetric reactions is one of the most important endeavours in current organic chemistry research.1 These ligands not only serve to transfer chirality to the reactants in the enantiodiscriminating step, but also modulate the steric and electronic behaviour of the metal to ensure high catalyst activity and productivity. The synthetically important2,3 Rh-catalyzed 1,4-addition of arylboronic acids to α,β-unsaturated ketones4 such as 2-cyclohexenone (1; Fig. 1a) provides an excellent illustration of this principle: the initial (in 1998) high yields and enantioselectivities with binap as the ligand (I; binap = 2,2-diphenylphosphino-1,1-binaphthyl)5 were improved while simultaneously increasing the reaction rate by a conceptually new ligand6 specially developed for this transformation (in 2003), Phbod (II, Phbod = 2,5-diphenylbicycloocta[2.2.2]-2,5-diene). Due to their higher activity,7 diene ligands have by and large replaced diphosphanes as the ligands of choice in the 1,4-addition and related arylation reactions.3,8 In 2008, Dorta et al. introduced atropisomeric biaryl-based chiral sulfoxides9,10 (analogues of binap; e.g., III) in the 1,4-addition reaction, which showed levels of performance that was considerably higher than diphosphanes and at par or even exceeding that of chiral dienes.
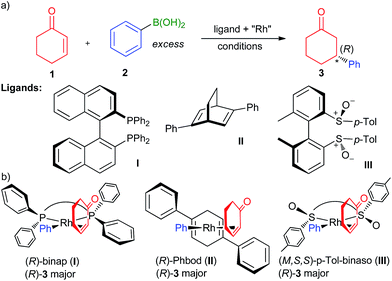 |
| | Fig. 1 (a) The asymmetric Rh-catalyzed 1,4-addition reaction and representative diphosphane,5 diene,12 and disulfoxide ligands10 showing high yields and enantioselectivities therein. (b) Pictorial models rationalizing the formation of the major enantiomer by avoiding steric repulsion with the forward-facing Ph group (diphosphane and diene) or dipole–dipole repulsion with the negatively charged oxygen atom (disulfoxide). | |
Explanation of the mechanism of action of chiral ligands goes hand in hand with discovery efforts. Based on the X-ray structure of [(binap)PdCl2] acquired in their investigation of a mechanistically related enantioselective Heck–Mizoroki reaction,11 Hayashi et al. proposed a pictorial model explaining the observed enantioselectivity by the avoidance of the steric clash of the ketone substituent and the large group (e.g., Ph in I5 and II12) on the ligand (Fig. 1b). Due to the excellent predictive power, visual appeal and intellectual elegance of this model, adaptations to different ligands and substrates have appeared in numerous of papers on Rh-catalyzed 1,4-additions in recent years.10,13,14 Computational modelling provides a powerful alternative to pictorial models because, being based on first principles, is not only theoretically sound but also permits quantitative evaluation of the enantioselectivity, which is much more important than simply predicting the reaction course. Recent DFT investigations of the origin of the enantioselectivity of bidentate phosphane,15,16 diene15,17,18 and disulfoxide19 ligands have shown that the Hayashi models quite accurately represent the structures of the enantiodiscriminating carborhodation transition state (CR-TS).
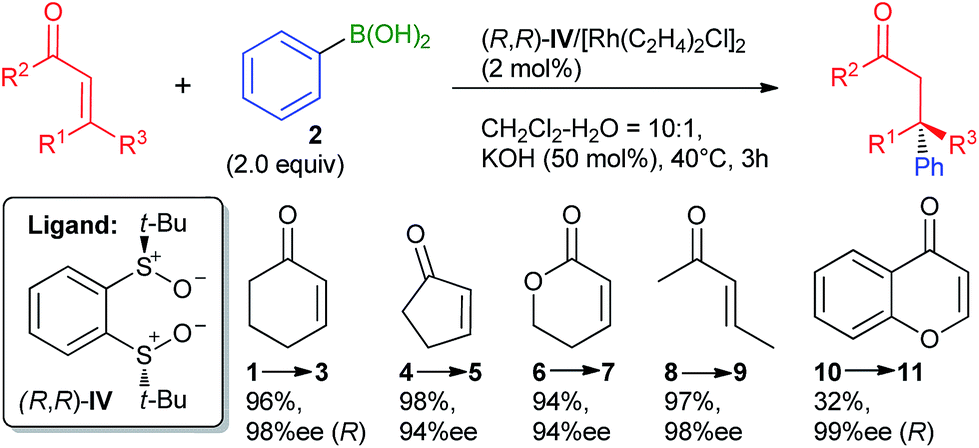
Until Dorta's work, chiral sulfoxide ligands had been used sporadically in various catalytic asymmetric reactions, usually showing uncompetitive levels of performance compared to other ligand classes.20 Dorta's discovery reignited the field and subsequent studies from various groups14,21,22 have solidified the position of chiral disulfoxide as excellent ligands for the Rh-catalyzed 1,4-addition. Particularly, a ligand developed by Liao et al.22 (IV; Scheme 1) is distinguished by uniformly high levels of enantioselectivity for a wide range of substrates achieved solely by the sulfoxide chirality. A computational study of the origin of the selectivity for ligand IV by density functional theory (DFT) is presented herein.
 |
| | Scheme 1 The previously published 1,4-arylation reaction mediated by (R,R)-IV/Rh.22 The absolute configuration of products 3 and 11 were assigned based on literature data. For 11, 5 mol% Rh and 3.0 equiv. 2 over 5 h were required. | |
Results and discussion
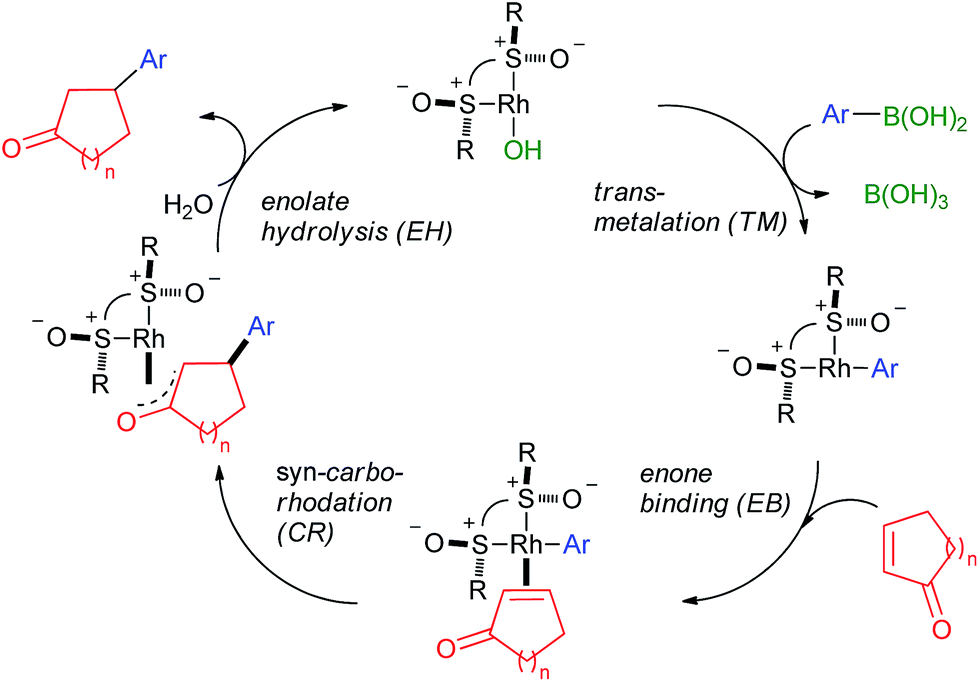
The accepted catalytic cycle23 of the 1,4-arylation (Scheme 1) comprises of transmetalation (TM), enone binding (EB), carborhodation (CR) and Rh-enolate hydrolysis (EH) steps, with the CR step being the one where the enantiodiscrimination takes place (Scheme 2). The reaction profile (Fig. 2a) for the EB + CR steps was calculated24 with the Perdew–Burke–Ernzerhof hybrid generalized gradient approximation (hGGA) functional containing 25% Hartree–Fock exchange (PBE0)25 combined with the full-electron, DFT-optimized DeGauss double-ζ + valence polarization basis set (DGDZVP)26 designed to give low basis set superposition error (BSSE). All structures were optimized in implicit CH2Cl2 by the integral equation formalism variant of the polarisable continuum model (IEFPCM)27 and the frequency calculations were performed at 40 °C (313.15 K) to (1) obtain the vibrational partition function hence ΔS and ΔG values at the reaction temperature; (2) verify the nature of the stationary points by the number of negative frequencies (0 for minima and 1 for TSs). 2-Cyclopentenone (4) was selected as the substrate due to its smaller size and flat shape. Starting from the key [((R,R)-IV)Rh-Ph] intermediate (12), two competing pathways, one leading to (R)-5 (R-pathway; 13–18) and the other – to (S)-5 (S-pathway; 19–23), need to be followed for computational characterization of the enantioselectivity. The reaction coordinate in this part of the cycle can be approximated by the Rh–C1 distance increasing from 2.003 Å in 12 to 4.334/4.010 Å in 18/23, respectively, and the C1–Cb decreasing from ∞ in 12 + 2 (the thermodynamic zero point) to 1.510/1.514 Å in 18/23, respectively. The reaction coordinate interacts with selected dihedral/inter-bond angles (4-atomic coordinates); this is the origin of the fundamentally important phenomenon that conformation strongly perturbs stationary point energies hence the selectivity of the reaction. During the EB stage (before CR), the main perturbing dihedral angle to consider is D1 (C1 → Rh → Ca → Cb; Fig. 2b); it can adopt up to 4 different values where local minima reside. D1 ∼ 180° (syn-EB) corresponds to the Rh-Ph and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond eclipsed in a way that places the Ph group near the ketone oxygen. Such orientation is unproductive for CR, and is expected to be the highest in energy. Therefore, such stationary points were not optimized. D1 ∼ 0° (anti-EB) corresponds to the only “eclipsed” orientation in which CR can take place. Two other “perpendicular” orientations are possible, close-EB and far-EB, in which the Rh-phenyl eclipses the ketone ring, or is situated opposite, respectively. In the R-pathway, all 3 conformers were found (13–15), with the far-EB (14; D1 = −102°, ΔG = −0.2 kcal mol−1) being the most stable and the anti-EB (15; D1 = −16°, ΔG = 8.6 kcal mol−1) being the least stable, both for the profile as a whole. The close-EB (13; D1 = 92°, ΔG = 2.7 kcal mol−1) is of intermediate stability as are the only 2 conformers found in the S-pathway, close-EB (19; D1 = −92°, ΔG = 0.7 kcal mol−1) and far-EB (20; D1 = 95°, ΔG = 3.7 kcal mol−1). All the EB intermediates are slightly distorted from the square-planar geometry typical for Rh(I) under the influence of the steric bulk of the neighbouring t-Bu group. However, alternative tetrahedral structures proved unstable. Note that in the Hayashi models (Fig. 1b), the anti-EB conformer is explicitly shown. However, our calculations for various ligands, including I,15 II,15,17 and now IV show that the EB step is highly variable, with the anti-EB conformers often being not the most stable, or not even existing at all.
C bond eclipsed in a way that places the Ph group near the ketone oxygen. Such orientation is unproductive for CR, and is expected to be the highest in energy. Therefore, such stationary points were not optimized. D1 ∼ 0° (anti-EB) corresponds to the only “eclipsed” orientation in which CR can take place. Two other “perpendicular” orientations are possible, close-EB and far-EB, in which the Rh-phenyl eclipses the ketone ring, or is situated opposite, respectively. In the R-pathway, all 3 conformers were found (13–15), with the far-EB (14; D1 = −102°, ΔG = −0.2 kcal mol−1) being the most stable and the anti-EB (15; D1 = −16°, ΔG = 8.6 kcal mol−1) being the least stable, both for the profile as a whole. The close-EB (13; D1 = 92°, ΔG = 2.7 kcal mol−1) is of intermediate stability as are the only 2 conformers found in the S-pathway, close-EB (19; D1 = −92°, ΔG = 0.7 kcal mol−1) and far-EB (20; D1 = 95°, ΔG = 3.7 kcal mol−1). All the EB intermediates are slightly distorted from the square-planar geometry typical for Rh(I) under the influence of the steric bulk of the neighbouring t-Bu group. However, alternative tetrahedral structures proved unstable. Note that in the Hayashi models (Fig. 1b), the anti-EB conformer is explicitly shown. However, our calculations for various ligands, including I,15 II,15,17 and now IV show that the EB step is highly variable, with the anti-EB conformers often being not the most stable, or not even existing at all.
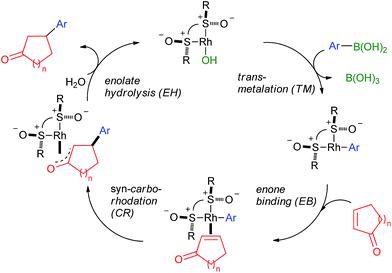 |
| | Scheme 2 The accepted catalytic cycle for the 1,4-arylation reaction with IV as the ligand (R = t-Bu). | |
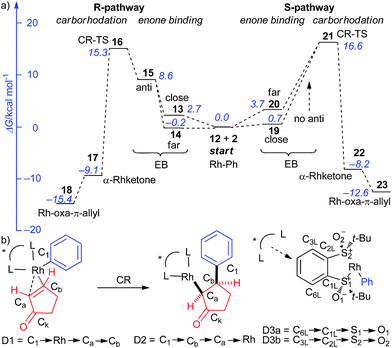 |
| | Fig. 2 (a) The reaction profile for the EB + CR stages of the catalytic cycle (Scheme 2) calculated at IEFPCM (CH2Cl2, 313.15 K)/PBE0/DGDZVP level of theory. (b) The main dihedral angles (D1–D3) that affect the reaction coordinate. | |
Upon CR, the D2 dihedral angle (C1 → Cb → Ca → Rh; Fig. 2b) becomes more chemically meaningful than D1. The R-pathway CR-TS (16; D1 = −23°, D2 = 27°, ΔG = 15.3 kcal mol−1) is lower in energy than the S-pathway CR-TS (21; D1 = −18°, D2 = 21°, ΔG = 16.6 kcal mol−1), correctly predicting (R)-5 as the major enantiomer in accord with the experiment. For the CR-TSs, we evaluated the effect of the t-Bu-sulfoxide rotation (dihedral angles D3a and D3b; Fig. 2b). As expected, the t-Bu group showed a very strong tendency to move away from the plane (D3 ∼ ±75°) of the o-benzylidene ring to avoid steric clash with the o-hydrogens, implying that ligand IV is not only bulky but also quite rigid. The pair of CR-TSs collapse to a pair of α-Rh ketones, in which the syn-periplanar orientation of Ph and [((R,R)-IV)Rh] moieties is largely preserved (R-pathway, 17, D2 = 5°, ΔG = −9.1 kcal mol−1; S-pathway, 22, D2 = 13°, ΔG = −8.2 kcal mol−1). A sliding motion of the Rh atom towards the ketone oxygen completes the 1,4-addition by forming a pair of Rh-oxa-π-allyl complexes while simultaneously relieving Ph/Rh torsional strain as evidenced by the increasing D2 values, leading to additional free energy gain (R-pathway, 18, D2 = 72°, ΔG = −9.1 kcal mol−1; S-pathway, 23, D2 = −29°, ΔG = −8.2 kcal mol−1). The (R)-product is thus both kinetically and thermodynamically preferred.
The EB step being mildly exergonic to endergonic implies that all of the EB conformers will be in rapid equilibrium among each other, and with the uncomplexed Rh–Ph species and enone. In such a case, the chirality of the product is determined solely by the energies of the CR-TSs that follow. This finding is common among representatives of 3 main ligand classes, diphosphanes (I),15 dienes (II)15,17 and disulfoxides (IV). Applying transition state theory to enantioselective catalysis28 shows that the enantiomeric excess can be calculated from the difference of the Gibbs energies of two competing, diastereomeric CR-TSs (ΔΔG‡ = ΔGCR-TS(R) − ΔGCR-TS(S)):
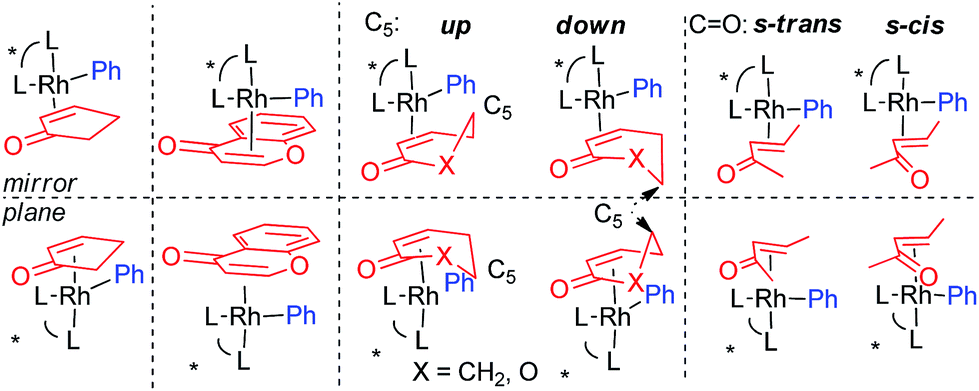
| |
 | (1) |
We calculated the required CR-TSs for all substrates in Scheme 1 with (R,R)-IV/Rh as the catalyst. The R- and S-pathways for substrates 1, 6 and 8 are additionally split as a consequence of the conformational specifics of these substrates (Fig. 3). The 6-member ring substrates 2-cyclohexenone (1) and 5,6-dihydro-2H-pyran-2-one (6) exist as a pair of inseparable chiral conformers, each of which can be distinguished by the Rh catalyst and C5 being on the same (“up”) or the opposite (“down”) side (middle). On the other hand, the linear (E)-3-penten-2-one (8) exists as a pair of diastereomeric s-trans and s-cis conformers (right). At IEFPCM (CH2Cl2, 313.15 K)/PBE0/DGDZVP level of theory, both conformers have equal energies. Therefore, for these substrates total of 4 CR-TSs each have to be calculated. 2-Cyclopentenone (4) or 4-chromenone (10), being flat, have no inherent conformations (left) and only 2 CR-TSs have to be considered. The comparison of the theoretical and predicted enantioselectivities (Table 1) showed that for all cyclic (Z)-olefin substrates, the major enantiomer was (R) whereas for the linear (E)-3-penten-2-one – (S). This suggests that that ligand (R,R)-IV exhibits the same sense of enantiodiscrimination as (R)-I and II and also indicates an excellent qualitative agreement between theory and experiment. Examination of the relative energies of the CR-TSs revealed that the chromenone lowest CR-TS (36) was 3.5 to 6.5 kcal mol−1 higher in energy than those for the smaller substrates (16, 24, 28 and 34, respectively). This indicates a lower rate of CR, which correlates well with the lower yield for this substrate under identical conditions (Scheme 1). Quantitatively, the IEFPCM/PBE0/DGDZVP model chemistry showed various degrees of accuracy. Interestingly, the theoretical enantioselectivities for the single-conformer cyclic substrates (4 and 10) were underestimated (by 0.8 and 1.2 kcal mol−1, respectively), those for the up/down double conformer cyclic substrates (1 and 6) were overestimated (by 0.8 and 0.5 kcal mol−1, respectively), and the one for the acyclic (E)-enone 8 was about right (0.2 kcal mol−1 underestimated). Inclusion of empirical correction for dispersion interactions, modelling of which is a traditional weakness of DFT, is often beneficial to improve quantitative agreement between experiment in theory.29 Therefore, we reoptimized all structures involved in the enantioselectivity prediction with SMD/M06 method. SMD30 is a recent parameterization variant of the PCM model including an improved set of van der Waals radii whereas M0631 is a recent, general-purpose hybrid-meta-GGA functional parameterized to satisfy a number of constraints also containing 25% Hartree–Fock. Both methods include dispersion-interaction corrections. The reaction barriers (ΔG‡) were systematically lowered with the SMD/M06 method (by 0.4–5.0 kcal mol−1). However, the accuracy in ΔΔG‡ hence predicted %ee improved considerably for the most problematic (for PCM/PBE0) substrates 4 and 10. On the other hand, the accuracy for the linear (E)-enone 8 decreased, but remained within the 1 kcal mol−1 range (along with all other substrates) recently proposed as an acceptable accuracy at the current state-of-the-art in DFT modelling of enantioselectivity.32 The origin of the discrepancies is unclear at present, but it underscores the significant challenges modelling of asymmetric catalysts pose to modern electronic structure theory. These findings are in line with our previous results for ligands I15 and II15,17 for substrates 1 and 4 as well.
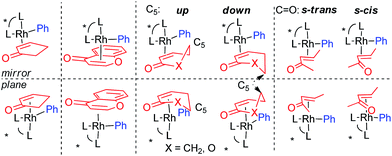 |
| | Fig. 3 Effect of substrate conformations on the reaction pathways and the number of the associated CR-TSs. | |
Table 1 Comparison of the calculated and experimental enantioselectivities with traditional PCM/PBE0 and dispersion-interaction-corrected SMD/M06 methods (both with DGDZVP basis set)
| s. m.a |
PCM (CH2Cl2, 313.15 K)/PBE0/DGDZVP |
SMD (CH2Cl2, 313.15 K)/M06/DGDZVP |
| ΔG (CR-TS)b, kcal mol−1 |
Enantioselectivityc |
ΔG (CR-TS)b, kcal mol−1 |
Enantioselectivityc |
| R-pathway |
S-pathway |
Theory |
Expt. |
R-pathway |
S-pathway |
Theory |
Expt. |
| S. m. = starting material (Scheme 1). Relative to ΔG(12) + ΔG(s.m.). Given in kcal mol−1 (%ee) and positive for the (R)-enantiomer. The two values were interconverted according eqn (1). ”Up”; “down”, respectively. ”s-cis”; “s-trans”, respectively. |
| 1 |
15.9 (24); 17.4 (25)d |
19.6 (26); 22.5 (27)d |
−3.7 (99.5) |
−2.9 (98) |
11.5 (24); 13.5 (25)d |
14.8 (26); 18.9 (27)d |
−3.3 (99.0) |
−2.9 (98) |
| 4 |
15.3 (16) |
16.6 (21) |
−1.3 (78) |
−2.2 (94) |
14.4 (16) |
16.2 (21) |
−1.8 (89) |
−2.2 (94) |
| 6 |
13.8 (28); 18.9 (29)d |
16.5 (30); 20.7 (31)d |
−2.7 (97.6) |
−2.2 (94) |
9.9 (28); 14.3 (29)d |
11.9 (30); 19.0 (31)d |
−2.0 (92) |
−2.2 (94) |
| 8 |
15.5 (32); 18.9 (33)e |
12.8 (34); 16.2 (35)e |
2.7 (−97.5) |
2.9 (−98) |
11.0 (32); 13.9 (33)e |
8.9 (34); 11.2 (35)e |
2.1 (−93) |
2.9 (−98) |
| 10 |
19.3 (36) |
21.4 (37) |
−2.1 (93) |
−3.3 (99) |
14.3 (36) |
17.2 (37) |
−2.9 (98) |
−3.3 (99) |
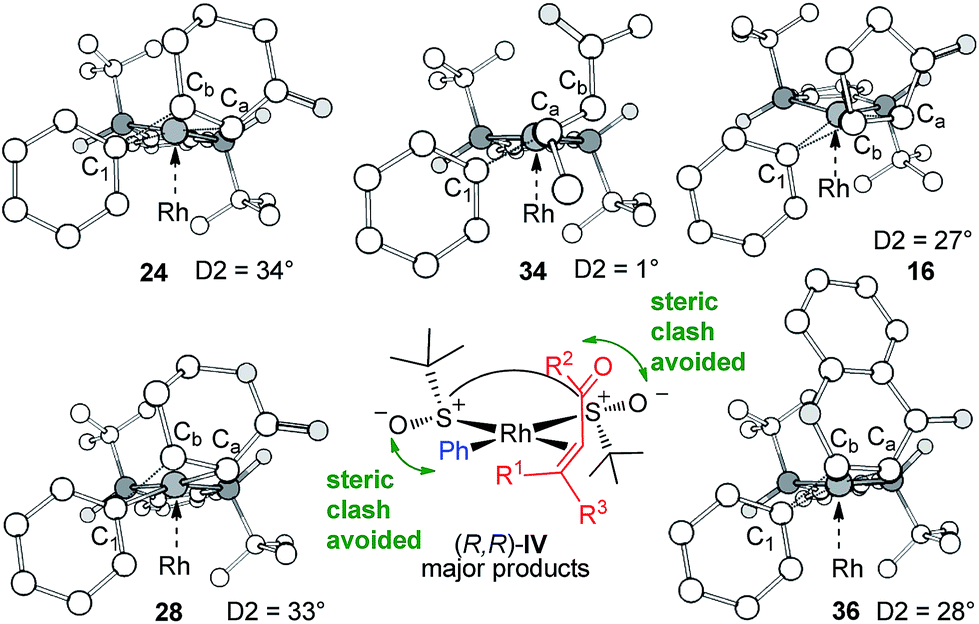
For all substrates, the CR-TSs adopt the distorted rectangular configuration characteristic for CR (migratory insertion) (Fig. 4). It is noteworthy that for ligand IV, the original premise behind the Hayashi model, namely the avoidance of the steric clash between the large ligand substituent (t-Bu in this case) and the ketone substituent on the opposite face of the plane remains valid, despite both t-Bu group and O atom pointing slightly backwards from the plane where the two S atoms lie. The high steric pressure exercised by the t-Bu groups is evident in the uneven, and at times quite large, degree of geometric distortion imposed on the key CR-TS structure encompassing C1, Ca, Cb, and Rh, and D2 dihedral angle these atoms form. The 6-member, non-planar substrates (1 and 6) are the most distorted (D2 = 34 and 33°, respectively) due to the steric clash between the C5–CH2 group sticking up, towards the t-Bu group on the opposite S atom (Fig. 4, left). On the other hand, the linear (E)-enone 8 fits very snugly between the two C2-symmetric t-Bu groups, resulting in a very small D2 angle (1°) (Fig. 4 middle, up). The situation with the flat cyclic substrates (4, 10) is intermediate (D2 = 27 and 28°, respectively; Fig. 4, right). Note that in all of the above structures, the Rh-phenyl group is pushed out of the Rh coordination plane by the bulky S-t-Bu substituent on the opposite face of the plane. This second instance of steric clash avoidance is not a feature of the original Hayashi model. Based, on the DFT results, we can propose a pictorial model (Fig. 4, middle, down) that embodies the idea of Liao et al., who concluded, based on the X-ray structure of the [((R,R)-IV)RhCl]2 complex, “the two tert-butyl groups provide an excellent stereoenvironment that may generate the high enantioselectivity in the 1,4-addition”.22
 |
| | Fig. 4 The optimized structures of the CR-TSs for all substrates (Table 1) leading to the major products. A Hayashi-type model consistent with both theoretical and experimental enantiodiscrimination is readily conceived from these structures. | |
In the above model, the ketone finds in a close proximity to the sulfoxide oxygen atom. Recall that the enantioselectivity of biaryl-derived sulfoxides was presumed to have been governed by the repulsion of the sulfoxide and ketone oxygens (Fig. 1b). Multiple weak interactions were found in the Atoms-In-Molecules (AIM)33 wavefunction analyzes (Fig. 5) of the two competing CR-TSs for 2-cyclopentenone (16 and 21), indicative of van der Waals interactions arising from steric hindrance. There is a weak interaction (ρb = 0.0050) in the major R-CR-TS (16), between the ketone oxygen and the sulfoxide oxygen that corresponds to the predicted dipole–dipole repulsion. However, it appears that the high steric pressure exercised by the t-Bu groups dominates. In that sense, ligand IV is similar to I, another high steric pressure15 ligand.
 |
| | Fig. 5 AIM plots for the two competing CR-TSs for 2-cyclopentenone. The critical points (CPs) are shown as follows: bond (B), red; ring (R), green; and cage (C), blue. Selected BCP electron densities (ρb) are also shown. | |
Conclusions
The enantioselective step in the 1,4-addition of phenylboronic acid to a variety Michael acceptors catalyzed Rh ligated with a disulfoxide ligand whose only source of chirality is the sulfur atoms was modelled by DFT (PCM/PBE0/DGDZVP level of theory). For all substrates, the predicted absolute configuration agreed with the experiment. However, the calculated %ee over or underestimated the experimental values. The high steric pressure exercised by the two t-Bu groups dominates over the weaker dipole–dipole repulsion between the ketone and sulfoxide O atom, and is the primary determinant of the enantioselectivity.
Acknowledgements
This work was supported by Hefei University of Technology, The Natural Sciences Foundation of Anhui Province (grant 11040606M35) and Wuhan University of Technology (China) and Institute of Materials Research and Engineering, A*STAR, (Singapore). The authors thank A*STAR Computational Resources Centre (Singapore) for the generous gift of computational time on the Axle, Fuji, Cirrus and Aurora HPC clusters.
Notes and references
- Comprehensive asymmetric catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Berlin, 1999 Search PubMed.
- H. J. Edwards, J. D. Hargrave, S. D. Penrose and C. G. Frost, Chem. Soc. Rev., 2010, 39, 2039–2105 RSC.
- G. Berthon and T. Hayashi, in Catalytic Asymmetric Conjugate Reactions, ed. A. Córdova, Wiley-VCH, 2010, pp. 1–70 Search PubMed.
- M. Sakai, H. Hayashi and N. Miyaura, Organometallics, 1997, 16, 4229–4231 CrossRef CAS.
- Y. Takaya, M. Ogasawara, T. Hayashi, M. Sakai and N. Miyaura, J. Am. Chem. Soc., 1998, 120, 5579–5580 CrossRef CAS.
-
(a) T. Hayashi, K. Ueyama, N. Tokunaga and K. Yoshida, J. Am. Chem. Soc., 2003, 125, 11508–11509 CrossRef CAS PubMed ; Simultaneously and independently, a C1-symmetrical chiral diene was developed for the Ir-catalyzed allylic substitution reaction;
(b) C. Fischer, C. Defieber, T. Suzuki and E. M. Carreira, J. Am. Chem. Soc., 2004, 126, 1628–1629 CrossRef CAS PubMed.
-
(a) A. Kina, H. Iwamura and T. Hayashi, J. Am. Chem. Soc., 2006, 128, 3904–3905 CrossRef CAS PubMed;
(b) A. Kina, Y. Yasunara, T. Nishimura, H. Iwamura and T. Hayashi, Chem.–Asian J., 2006, 1, 707–711 CrossRef CAS PubMed.
-
(a) P. Tian, H.-Q. Dong and G.-Q. Lin, ACS Catal., 2012, 2, 95–119 CrossRef CAS;
(b) T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829–2844 CrossRef CAS PubMed;
(c) K. Fagnou and M. Lautens, Chem. Rev., 2003, 103, 169–196 CrossRef CAS PubMed.
- R. Mariz, X. Luan, M. Gatti, A. Linden and R. Dorta, J. Am. Chem. Soc., 2008, 130, 2172–2173 CrossRef CAS PubMed.
- J. J. Bürgi, R. Mariz, M. Gatti, E. Drinkel, X. Luan, S. Blumentritt, A. Linden and R. Dorta, Angew. Chem., Int. Ed., 2009, 48, 2768–2771 CrossRef PubMed.
- F. Ozawa, A. Kubo, Y. Matsumoto, T. Hayashi, K. Nishioka, K. Yanagi and K. Moriguchi, Organometallics, 1993, 12, 4188–4196 CrossRef CAS.
- G. Berthon-Gelloz and T. Hayashi, J. Org. Chem., 2006, 71, 8957–8960 CrossRef CAS PubMed.
-
(a) T. Nishimura, A. Noishiki, C. C. Tsui and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 5056–5059 CrossRef CAS PubMed;
(b) K. Sasaki, T. Nishimura, R. Shintani, E. A. B. Kantchev and T. Hayashi, Chem. Sci., 2012, 3, 1278–1283 RSC;
(c) D. W. Low, G. Pattison, M. D. Wieczysty, G. H. Churchill and H. W. Lam, Org. Lett., 2012, 14, 2548–2551 CrossRef CAS PubMed;
(d) Y. Luo, H. B. Hepburn, N. Chotsaeng and H. W. Lam, Angew. Chem., Int. Ed., 2012, 51, 8309–8313 CrossRef CAS PubMed;
(e) Y. Luo, A. J. Carnell and H. W. Lam, Angew. Chem., Int. Ed., 2012, 51, 6762–6766 CrossRef CAS PubMed;
(f) J. Csizmadiová, M. Mečiarová, E. Rakovský, B. Horváth and R. Šebesta, Eur. J. Org. Chem., 2011, 6110–6116 CrossRef;
(g) A. Saxena and H. W. Lam, Chem. Sci., 2011, 2, 2326–2331 RSC;
(h) R. Shintani and T. Hayashi, Org. Lett., 2011, 13, 350–352 CrossRef CAS PubMed;
(i) R. Shintani, M. Takeda, Y.-T. Soh, T. Ito and T. Hayashi, Org. Lett., 2011, 13, 2977–2979 CrossRef CAS PubMed;
(j) R. Shintani, S. Isobe, M. Takeda and T. Hayashi, Angew. Chem., Int. Ed., 2010, 49, 3795–3798 CrossRef CAS PubMed;
(k) G. Pattison, G. Piraux and H. W. Lam, J. Am. Chem. Soc., 2010, 132, 14373–14375 CrossRef CAS PubMed;
(l) T. Nishimura, H. Makino, M. Nagaosa and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 12865–12867 CrossRef CAS PubMed;
(m) C. Shao, H.-J. Yu, N.-Y. Wu, C.-G. Feng and G.-Q. Lin, Org. Lett., 2010, 12, 3820–3823 CrossRef CAS PubMed;
(n) T. Gendrineau, O. Chuzel, H. Eijsberg, J.-P. Genet and S. Darses, Angew. Chem., Int. Ed., 2008, 47, 7669–7672 CrossRef CAS PubMed;
(o) K. Okamoto, T. Hayashi and V. H. Rawal, Org. Lett., 2008, 10, 4378–4389 Search PubMed;
(p) F. Läng, F. Breher, D. Stein and H. Grützmacher, Organometallics, 2005, 24, 2997–3007 CrossRef;
(q) N. Tokunaga, Y. Otomaru, K. Okamoto, K. Ueyama, R. Shintani and T. Hayashi, J. Am. Chem. Soc., 2004, 126, 13584–13585 CrossRef CAS PubMed.
- Q. Chen, C. Chen, F. Guo and W. Xia, Chem. Commun., 2013, 49, 6433–6435 RSC.
- H.-L. Qin, X.-Q. Chen, Y.-Z. Huang and E. A. B. Kantchev, Chem.–Eur. J., 2014, 20, 12982–12987 CrossRef CAS PubMed.
-
(a) T. Korenaga, R. Maenishi, K. Hayashi and T. Sakai, Adv. Synth. Catal., 2010, 352, 3247–3254 CrossRef CAS;
(b) P. Mauleon, I. Alonso, M. Rodriguez Rivero and J. C. Carretero, J. Org. Chem., 2007, 72, 9924–9935 CrossRef CAS PubMed;
(c) T. Itoh, T. Mase, T. Nishikata, T. Iyama, H. Tachikawa, Y. Kobayashi, Y. Yamamoto and N. Miyaura, Tetrahedron, 2006, 62, 9610–9621 CrossRef CAS PubMed.
- E. A. B. Kantchev, Chem. Sci., 2013, 4, 1864–1875 RSC.
-
(a) S. Gosiewska, J. A. Raskatov, R. Shintani, T. Hayashi and J. M. Brown, Chem.–Eur. J., 2012, 80–84 CrossRef CAS PubMed;
(b) E. A. B. Kantchev, Chem. Commun., 2011, 47, 10969–10971 RSC.
-
(a) A. Poater, F. Ragone, R. Mariz, R. Dorta and L. Cavallo, Chem.–Eur. J., 2010, 16, 14348–14353 CrossRef CAS PubMed;
(b) R. Mariz, A. Poater, M. Gatti, E. Drinkel, J. J. Bürgi, X.-J. Luan, S. Blumentritt, A. Linden, L. Cavallo and R. Dorta, Chem.–Eur. J., 2010, 16, 14335–14347 CrossRef CAS PubMed.
-
(a) E. Ichikawa, M. Suzuki, K. Yabu, M. Albert, M. Kanai and M. Shibasaki, J. Am. Chem. Soc., 2004, 126, 11808–11809 CrossRef CAS PubMed;
(b) R. Tokinoh, M. Sodeoka, K. Aoe and M. Shibasaki, Tetrahedron Lett., 1995, 44, 8035–8038 CrossRef;
(c) N. Khiar, I. Fernández and F. Alaudia, Tetrahedron Lett., 1993, 34, 123–126 CrossRef CAS;
(d) B. R. James and R. S. McMillan, Can. J. Chem., 1977, 55, 3927–3932 CrossRef CAS.
-
(a) N. Khiar, A. Salvador, V. Valdivia, A. Chelouan, A. Alcudia, E. Álvarez and I. Fernández, J. Org. Chem., 2013, 78, 6510–6521 CrossRef CAS PubMed;
(b) F. Han, G. Chen, X. Zhang and J. Liao, Eur. J. Org. Chem., 2011, 2928–2931 CrossRef CAS;
(c) P. K. Dornan, P. L. Leung and V. M. Dong, Tetrahedron, 2011, 67, 4378–4384 CrossRef CAS PubMed;
(d) Q.-A. Chen, X. Dong, M.-W. Chen, D.-S. Wang, Y.-G. Zhou and Y.-X. Li, Org. Lett., 2010, 12, 1928–1931 CrossRef CAS PubMed.
- J. Chen, J. Chen, F. Lang, X. Zhang, L. Cun, J. Zhu, J. Deng and J. Liao, J. Am. Chem. Soc., 2010, 132, 4552–4553 CrossRef CAS PubMed.
- T. Hayashi, M. Takahashi, Y. Takaya and M. Ogasawara, J. Am. Chem. Soc., 2002, 124, 5052–5058 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3685–3868 CrossRef CAS ; errata J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef.
- N. Godbout, D. R. Salahub, J. Andzelm and E. Wimmer, Can. J. Chem., 1992, 70, 560–571 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed and refs. cited therein.
- D. Balcells and F. Maseras, New J. Chem., 2007, 31, 333–343 RSC.
-
(a) S. Ehrlich, J. Moellmann and S. Grimme, Acc. Chem. Res., 2013, 46, 916–926 CrossRef CAS PubMed;
(b) S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 211–228 CrossRef CAS;
(c) T. Schwabe and S. Grimme, Acc. Chem. Res., 2008, 41, 569–579 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- A. Armstrong, R. A. Boto, P. Dingwall, J. Contreras-García, M. J. Harvey, N. J. Mason and H. S. Rzepa, Chem. Sci., 2014, 5, 2057–2071 RSC.
- For a general introduction, see: R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Clarendon Press, Oxford, UK, 1990. The AIM calculations were performed with AIMAll software: T. A. Keith, TK Gristmill Software, Overland Park KS, USA, 2014 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Cartesian coordinates, energies and first 3 frequencies for all stationary points and IRC plots for selected transition states. See DOI: 10.1039/c4ra13012j |
| ‡ Former address: Institute of Materials Research and Engineering, 3 Research Link, Singapore 117602. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.