
Pyrroloquinoline quinone maintains redox activity when bound to a DNA aptamer†
Abstract
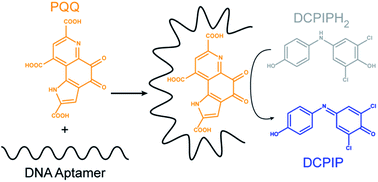
We have identified by in vitro selection DNA aptamers for the redox cofactor pyrroloquinoline quinone (PQQ). Using a spectroscopic assay, we determined PQQ maintains its redox properties when bound to the DNA aptamers. These complexes could find potential use as biocatalysts when direct electrical communication with electrode surfaces is desirable.
 Please wait while we load your content...
Please wait while we load your content...

