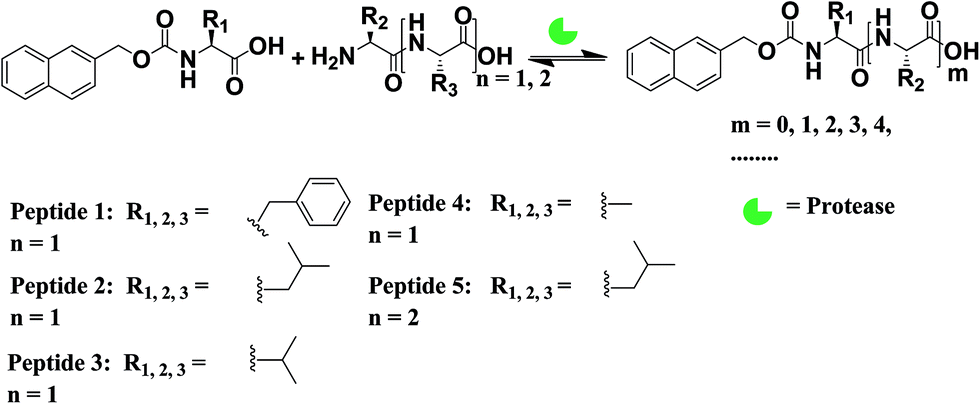
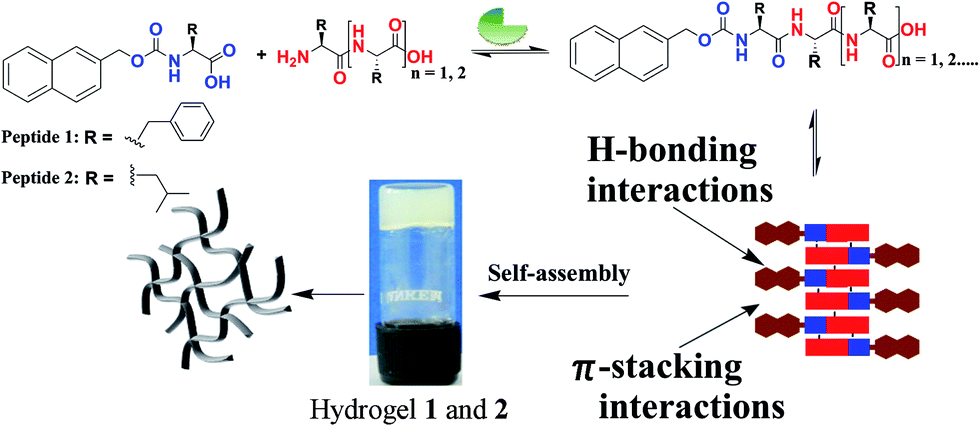
DOI:
10.1039/C4RA09490E
(Paper)
RSC Adv., 2015,
5, 1529-1537
Exploring thermodynamically downhill nanostructured peptide libraries: from structural to morphological insight†
Received
30th August 2014
, Accepted 18th November 2014
First published on 18th November 2014
Abstract
Here, we report the biocatalytic evolution of Nmoc (naphthalene-2-methoxycarbonyl)-capped dynamic combinatorial peptide libraries in the hydrogel state. Our approach is to use a biocatalyst, which can bring up the peptide self-assembly via synthesis and in situ self-organization of peptide oligomers under physiological conditions. The enzyme drives the amplification of Nmoc-capped peptide oligomers and leads to the generation of dynamic combinatorial libraries under physiological conditions via a reverse hydrolysis reaction. The enzyme permits reversible peptide synthesis as well as peptide hydrolysis reactions, which generate a preferred nanostructured component through peptide self-assembly. Nmoc-F/FF and Nmoc-L/LL systems have been used successfully to generate Nmoc-F3 and Nmoc-L5 as preferred components in the dynamic peptide libraries, which form helical nanostructures. The control experiment with a Nmoc-L/LLL system depicts the selection and preferred formation of a Nmoc-L5 library member via self-assembly. The library components are analysed by reverse phase high performance liquid chromatography (RP-HPLC) and mass spectrometry. The self-assembled nanomaterials are studied by rheology, fluorescence and time correlated single photon counting (TCSPC) spectroscopy. The secondary structure of the peptide components are analysed by FT-IR and circular dichroism (CD) spectroscopy. The self-assembled nanostructures are imaged by atomic force microscopy (AFM) and transmission electron microscopy (TEM).
Introduction
Self-assembled amphiphilic peptides1–3 have been used in constructing nanostructured biomaterials due to their potential applications in drug delivery4–7 and biomedical applications.8,9 Self-assembly is the result of non-covalent interactions including π–π stacking, hydrogen bonding and hydrophobic interactions of self-assembled molecules. Low molecular weight amphiphilic small molecules can entrap >99% of water and form a supramolecular hydrogel.10–13 A hydrogel is composed of complex three dimensional nanofibrillar networks,14,15 which can entrap large volumes of water. Small peptide amphiphiles are used to design self-supporting hydrogels with more ordered nanostructures.16 Enzyme catalysed hydrogelation is more promising due to its broad biomedical and nanotechnological applications.17–19
Over the last few years, dynamic combinatorial libraries (DCLs)20–22 of small molecules have emerged as powerful tools to evolve complex nanostructures. The library components undergo continuous bond breaking and making under thermodynamic control, which allows the system to reach an equilibrium state. In a DCL, the inter-conversion of library members occurs through a reversible chemical reaction, which can introduce new covalent23–25 and non-covalent interactions. Self-assembly driven dynamic libraries are generated under various external stimuli, including metal ions,26 chemical templates,27 temperature28 and enzymes.29 External stimuli can drive the library members to reorganize in a particular fashion in order to minimize the total free energy of the system. Typically, enzymes are used to hydrolyse the polymers for controlled drug delivery applications.30 Enzymes, mainly proteases, are also used to exploit the peptide self-assembly via reverse hydrolysis reactions.31–33 Self-assembling peptide hydrogel systems are also achieved via lipase catalysed reverse hydrolysis reactions.34,35 Steroidal motifs were used to generate hydrazone based DCLs.36 Otto et al. described peptide based dynamic combinatorial libraries based on a disulfide exchange reaction.37 Lehn et al. envisaged a constitutional dynamic hydrogel based on the self-assembly and reversible connection of library components.38 The stabilization of a library component among the library members depends on the formation of stable assemblies or aggregates. In the pool of enzymatic chemical reactions, the self-assembly of a library component shifts the equilibrium towards its formation. This results in the amplification of a preferred self-assembled component over the less or dis-assembled components. Previously, the N-terminus of amino acids or peptides was functionalized with an aromatic fluorenyl methoxycarbonyl (Fmoc) group,39,40 which plays a crucial role in the self-assembly process. Ulijn et al. reported a fully reversible self-assembly of small low molecular weight peptides under thermodynamic control.32 Palocci et al. described the use of microbial lipases for the synthesis of Fmoc-tripeptide results in the formation of a gel.34 Recently, we have explored π-stacked β-sheet dynamic peptide libraries through the hydrolysis of peptides, which are catalysed by an enzyme.41
In here, we explore enzyme catalysed dynamic peptide libraries, in which a protease enzyme can bring up the self-assembly of peptides through the reverse hydrolysis of aromatic Nmoc (naphthalene-2-methoxycarbonyl)-capped amino acids and dipeptides. Naphthalene-based peptides are an interesting class of low molecular weight gelators, which have been used in cell culture,42 energy transfer43 and drug delivery5 applications. Here, we report enzymatic reactions of Nmoc-amino acids with a variety of dipeptides or a tripeptide, which lead to the generation of dynamic combinatorial libraries and consequently the self-assembly can drive the amplification of a library component among the library members. The thermodynamic stabilization of a preferred library component in a gel phase medium forms a complex nanostructure.
Results and discussion
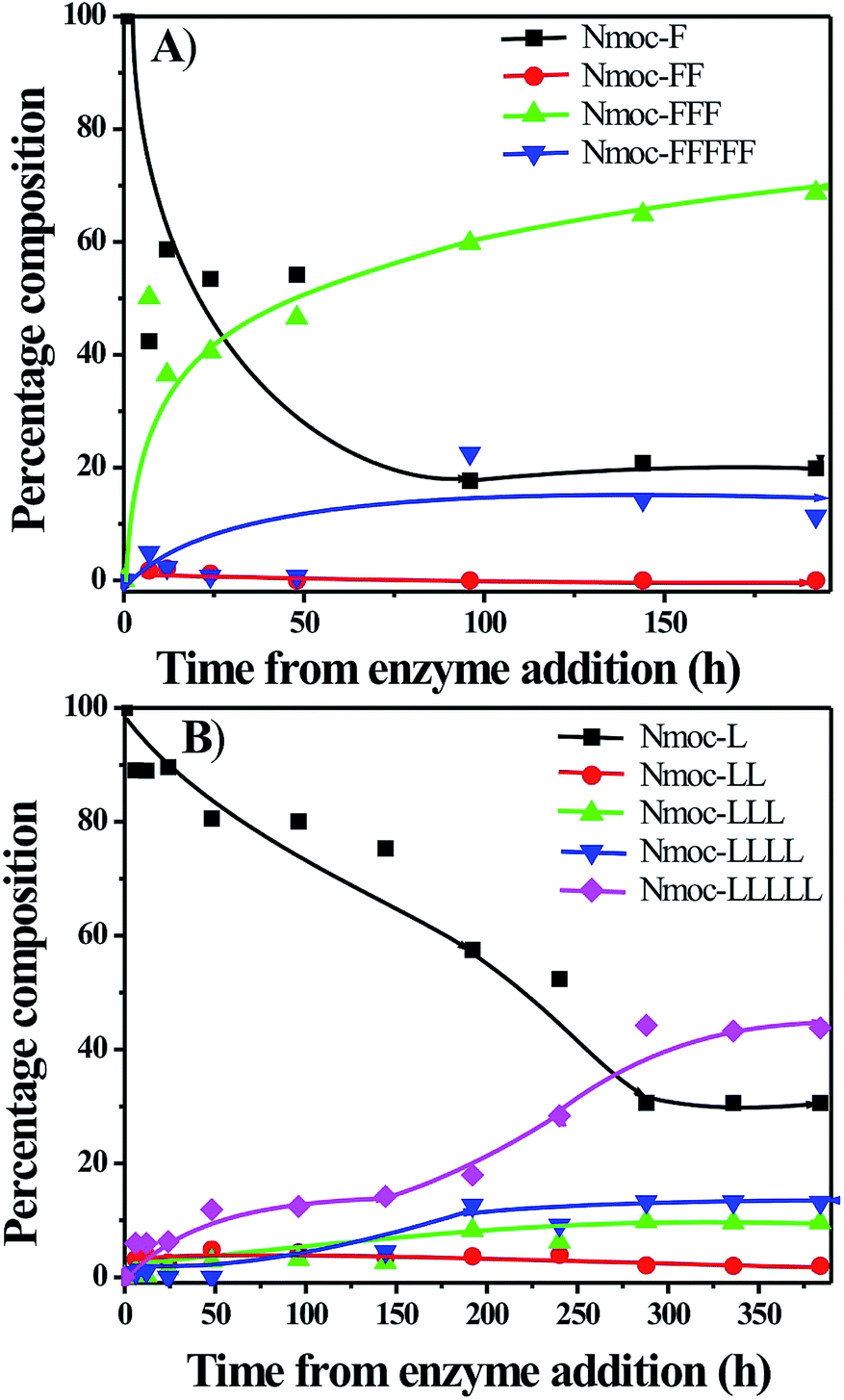
Nmoc-amino acids (Phe: F, Leu: L, Val: V and Ala: A), dipeptides (FF, LL, VV and AA) and a tripeptide (LLL) were synthesized to generate dynamic combinatorial libraries in an aqueous medium (Schemes 1, S1–S3†). Dynamic combinatorial chemistry is a powerful tool to generate a complex nanostructure in a supramolecular hydrogel. We have carried out an enzyme catalysed reverse hydrolysis reaction with Nmoc-protected amino acids and dipeptides to generate dynamic combinatorial libraries. Nmoc-F/FF (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60 mmol L−1) and Nmoc-L/LL (20:60 mmol L−1) were dissolved in 2 mL phosphate buffer (100 mmol, pH ∼ 8) in each case. The milky suspension was mixed to make a homogeneous solution and kept undisturbed at room temperature. The self-assembly through an enzymatic chemical reaction was observed (Fig. 1), when the enzyme, thermolysin (1 mg mL−1), was added to the respective solution (Table 1). The self-assembled self-supporting hydrogelation was observed within 5 min for the Nmoc-F/FF system with 40% conversion of Nmoc-F3. However, the Nmoc-L/LL system took around 36 h to form a self-assembled hydrogel. The HPLC analysis exhibits the formation of Nmoc-based peptide oligomers, which clearly indicates the selectivity of the enzyme towards a reverse hydrolysis reaction. The newly synthesized library components were confirmed by HPLC and ESI-MS (Fig. S1–S12†). Nmoc-F3 was formed as a major component in the self-assembled Nmoc-F/FF hydrogel system with 71% conversion, whereas Nmoc-L5 formed predominantly with 44% conversion for the self-assembled Nmoc-L/LL hydrogel system (Fig. 2). In here, Nmoc-F3, of the Nmoc-F/FF system, and Nmoc-L5, of the Nmoc-L/LL system, are the self-assembled components over the other non-assembled components. Our enzyme catalysed amplification of Nmoc-L/LL and Nmoc-F/FF results are very similar to the results32 of the Fmoc-L/LL and Fmoc-F/FF34,35 systems.
:60 mmol L−1) and Nmoc-L/LL (20:60 mmol L−1) were dissolved in 2 mL phosphate buffer (100 mmol, pH ∼ 8) in each case. The milky suspension was mixed to make a homogeneous solution and kept undisturbed at room temperature. The self-assembly through an enzymatic chemical reaction was observed (Fig. 1), when the enzyme, thermolysin (1 mg mL−1), was added to the respective solution (Table 1). The self-assembled self-supporting hydrogelation was observed within 5 min for the Nmoc-F/FF system with 40% conversion of Nmoc-F3. However, the Nmoc-L/LL system took around 36 h to form a self-assembled hydrogel. The HPLC analysis exhibits the formation of Nmoc-based peptide oligomers, which clearly indicates the selectivity of the enzyme towards a reverse hydrolysis reaction. The newly synthesized library components were confirmed by HPLC and ESI-MS (Fig. S1–S12†). Nmoc-F3 was formed as a major component in the self-assembled Nmoc-F/FF hydrogel system with 71% conversion, whereas Nmoc-L5 formed predominantly with 44% conversion for the self-assembled Nmoc-L/LL hydrogel system (Fig. 2). In here, Nmoc-F3, of the Nmoc-F/FF system, and Nmoc-L5, of the Nmoc-L/LL system, are the self-assembled components over the other non-assembled components. Our enzyme catalysed amplification of Nmoc-L/LL and Nmoc-F/FF results are very similar to the results32 of the Fmoc-L/LL and Fmoc-F/FF34,35 systems.
 |
| | Scheme 1 Enzyme catalysed reverse hydrolysis reaction of Nmoc-amino acids with di- and tri-peptides, which evolve dynamic peptide libraries. | |
 |
| | Fig. 1 Enzyme catalysed reverse hydrolysis reaction of Nmoc-amino acids and dipeptides. The schematic representation describes the enzyme catalysed evolution of dynamic peptide libraries of the Nmoc-F/FF and Nmoc-L/LL systems, and the enzyme catalysed amplification of self-assembled Nmoc-F3, of the Nmoc-F/FF system, and Nmoc-L5, of the Nmoc-L/LL system, to form nanostructures in a gel phase. | |
Table 1 Product formation upon a biocatalytic reverse hydrolysis reactiona
| Entry |
Starting |
Enzyme |
Nmoc-X% |
Nmoc-X2% |
Nmoc-X3% |
Nmoc-X4% |
Nmoc-X5% |
| Nmoc: naphthalene-2-methoxycarbonyl, X = phenylalanine (entry 1); X = leucine (entry 2 and 5); X = valine (entry 3) and X = alanine (entry 4). |
| 1 |
Nmoc-F + FF |
1 mg mL−1 |
18.56 |
0 |
71.29 |
3.77 |
6.39 |
| 2 |
Nmoc-L + LL |
1 mg mL−1 |
30.82 |
2.02 |
9.58 |
13.30 |
44.28 |
| 3 |
Nmoc-V + VV |
1 mg mL−1 |
100 |
0 |
0 |
0 |
0 |
| 4 |
Nmoc-A + AA |
1 mg mL−1 |
100 |
0 |
0 |
0 |
0 |
| 5 |
Nmoc-L + LLL |
1 mg mL−1 |
74.81 |
0.12 |
0.1 |
3.68 |
21.29 |
 |
| | Fig. 2 Time course of the conversion of the peptide oligomers in dynamic combinatorial libraries of the (A) Nmoc-F/FF system and the (B) Nmoc-L/LL system. | |
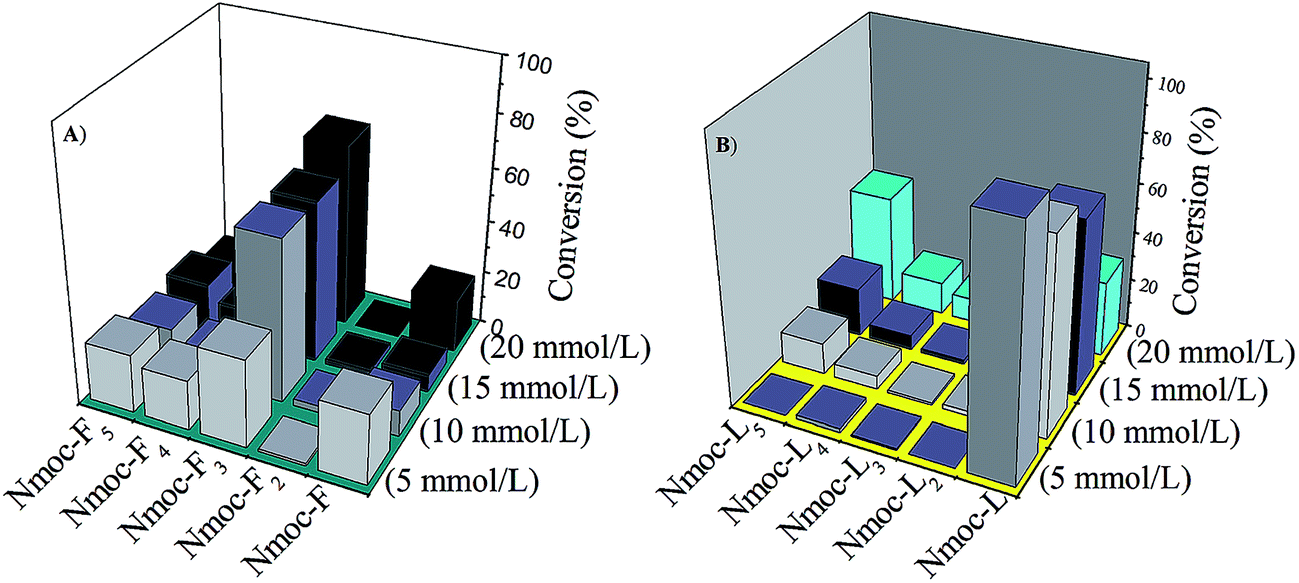
The enzyme catalysed newly synthesised Nmoc-F3 and Nmoc-L5 oligomers self-assembled in an aqueous medium and turned to hydrogels. However, to evaluate the amplification of the self-assembled components in the dynamic libraries, the Nmoc-F/FF and Nmoc-L/LL systems were studied at different concentrations. The Nmoc-F/FF system at a lower concentration (5:15 mmol L−1) showed a random distribution of library components and all the library components remained in solution form while the Nmoc-L/LL system didn’t show peptide conversion upon enzyme addition at a lower (5:15 mmol L−1) concentration (Fig. 3). Moreover, after increasing the Nmoc-amino acid concentration from 5 to 20 mmol L−1, Nmoc-F3 and Nmoc-L5 were formed as a preferred component for the Nmoc-F/FF and Nmoc-L/LL systems, respectively. The Nmoc-L/LL system formed Nmoc-L5 by subsequent coupling of dipeptide LL with Nmoc-L, which results in the self-assembly. However, the preferred components or library members self-organize or self-assemble at their minimum energy state. This was clearly demonstrated by a control experiment with the Nmoc-L/LLL system. In this case, Nmoc-L4 was generated by the Nmoc-L/LLL system with 52% conversion, which collapsed over time and Nmoc-L5 dominated in the DCL pool (Fig. 4). Thus, the self-assembly provides a driving force for the generation and self-selection of peptides via the breaking and making of peptide bonds in protease triggered dynamic combinatorial libraries. The Nmoc-L/LLL system proves that the self-assembly drives the formation of the thermodynamically stable product among the library members (Fig. 5). The Nmoc-V/VV and Nmoc-A/AA systems were unable to form a hydrogel and no product conversion was observed.
 |
| | Fig. 3 The formation of peptide oligomers from an enzyme catalysed dynamic library of the (A) Nmoc-F/FF and (B) Nmoc-L/LL systems at different concentrations. | |
 |
| | Fig. 4 Real time conversion of peptide oligomers in a dynamic peptide library of the Nmoc-L/LLL system. | |
 |
| | Fig. 5 Free energy profile diagram of the enzyme catalysed dynamic peptide libraries. The Nmoc-L/LL system forms Nmoc-L5 via amide bond formation, which subsequently self-assembles to form nanofibrillar structures. Nmoc-L/LLL initially forms Nmoc-L4, which leads to the formation of the thermodynamically favoured Nmoc-L5, followed by the self-assembly. | |
In order to generate a more complex system and to evolve the unexpected library member, Nmoc-L was mixed with LL, VV, AA and GG dipeptides. The library members, Nmoc-L5 (20%), Nmoc-LLVV (19%) and Nmoc-LLLVV (12%), were formed after 10 days (Fig. 6). In here, two different major peptide oligomers, Nmoc-L5 and Nmoc-LLVV, were unable to form a self-supporting hydrogel, indicating no supramolecular arrangement among different peptide oligomers. This complex Nmoc-L/LL/VV/AA/GG system led to the formation of more complex reaction products and was unable to self-assemble due to a random distribution of library components. Our results of the Nmoc-L/LL system showed that Nmoc-L can couple with LL, but was unable to couple with the other dipeptides, VV, AA and GG. However, the synthesized Nmoc-LL and Nmoc-LLL coupled with VV and formed the Nmoc-LLVV and Nmoc-LLLVV peptides (Fig. S13–S15†). Thus, our result indicates that the protease catalysed amplification drives towards a preferred product formation (Fig. 6).
 |
| | Fig. 6 (A) High performance liquid chromatography (HPLC) analysis of the Nmoc-L/LL, VV, AA and GG systems, which form a dynamic library of peptides upon treatment with thermolysin after 8 days. (B) Time course of the conversion of peptide oligomers in dynamic combinatorial libraries of the Nmoc-L/LL, VV, AA and GG systems. | |
The peptide self-assembly in the DCLs is influenced by various non-covalent interactions. The hydrogen bonding interactions of amide bonds play a crucial role in the self-assembly process, which is responsible for peptide secondary structures. Therefore, FTIR is used to monitor the secondary structure of peptides. The self-assembled peptides acquire a particular secondary structure, which can be elucidated based on an amide I region (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the amide bond) in FTIR. The oxygen atom of CO of the amide bonds acts as a donor in the hydrogen bonding interactions in the peptide self-assembly. The self-assembled structure from the Nmoc-F/FF and Nmoc-L/LL systems showed characteristic peaks in the amide I region. A preferred library component, Nmoc-F3, was responsible for the formation of a self-supporting hydrogel of the Nmoc-F/FF system. The peptide, Nmoc-F3, showed peaks at 1636 cm−1 and 1689 cm−1 in FTIR, which indicate an antiparallel β-sheet structure in the self-assembled hydrogel state44–46 (Fig. 7). Moreover, Nmoc-L5 was obtained preferentially from the Nmoc-L/LL system, which was responsible for the hydrogel formation. The preferred library member, Nmoc-L5, exhibited a characteristics peak at 1636 cm−1 in the amide I region (Fig. 7). The presence of the peak indicates that two peptide backbones are held together by hydrogen bonding interactions, which is responsible for β-sheet conformation. The self-organization at the molecular level establishes a β-sheet structure, which forms a helical structure at the higher level molecular self-assembly.
O stretching vibration of the amide bond) in FTIR. The oxygen atom of CO of the amide bonds acts as a donor in the hydrogen bonding interactions in the peptide self-assembly. The self-assembled structure from the Nmoc-F/FF and Nmoc-L/LL systems showed characteristic peaks in the amide I region. A preferred library component, Nmoc-F3, was responsible for the formation of a self-supporting hydrogel of the Nmoc-F/FF system. The peptide, Nmoc-F3, showed peaks at 1636 cm−1 and 1689 cm−1 in FTIR, which indicate an antiparallel β-sheet structure in the self-assembled hydrogel state44–46 (Fig. 7). Moreover, Nmoc-L5 was obtained preferentially from the Nmoc-L/LL system, which was responsible for the hydrogel formation. The preferred library member, Nmoc-L5, exhibited a characteristics peak at 1636 cm−1 in the amide I region (Fig. 7). The presence of the peak indicates that two peptide backbones are held together by hydrogen bonding interactions, which is responsible for β-sheet conformation. The self-organization at the molecular level establishes a β-sheet structure, which forms a helical structure at the higher level molecular self-assembly.
 |
| | Fig. 7 FTIR spectra of self-assembled peptides Nmoc-F3 and Nmoc-L5 obtained from Nmoc-F/FF at 8 days and Nmoc-L/LL at 16 days. The peaks in the amide I region indicate a β-sheet conformation in the gel phase. | |
Circular Dichroism (CD) measurements were also performed to better assess the nature of the secondary structure of the self-assembled peptides. The CD spectrum of the Nmoc-F/FF system shows characteristic negative peaks at 222 nm and 212 nm, which are attributed to the n–π* transition of the CONH groups of peptide Nmoc-F3. This signature is analogous to the reported CD signature of peptides adopting a typical helical conformation (Fig. 8).47–49 The helical conformation of peptide nanostructures is consistent with the atomic force microscopy results. The CD analysis of the Nmoc-L/LL system interestingly shows β-sheet and helical structures. A characteristic negative peak was observed at 219 nm and a positive peak appeared at 203 nm (Fig. 8). This observation indicates that the gelator molecules self-assembled50–54 into β-sheet and helical structures through extensive hydrogen-bonding interactions.
 |
| | Fig. 8 (A) Circular dichroism (CD) spectra of hydrogel 1 at 8 days and hydrogel 2 at 16 days, which reveal the formation of β-sheet and helical structures through extensive hydrogen-bonding interactions. | |
A fluorescence study of hydrogels 1 and 2 was carried out to get more insight into the molecular arrangement of the self-assembled peptides. UV-Vis absorbance spectra were recorded to know the exact excitation wavelength of hydrogels 1 and 2 (Fig. S16†). An aromatic π-stacking interaction55–57 is known to be the driving force behind the self-assembly of aromatic peptide amphiphiles. Aromatic Nmoc (naphthalene-2-methoxycarbonyl)-protected peptide hydrogels 1 and 2 exhibit strong emission peaks at 346 nm and 341 nm, respectively (Fig. S17†). The emission peaks at 346 nm and 341 nm for the hydrogels are attributed to the π-stacking interaction of the aromatic naphthalene rings in the hydrogel state. An emission at 465 nm has been observed for hydrogel 1. The emission at 465 nm indicates an excimer formation of the Nmoc groups in the gel phase. The emission peaks for both hydrogels indicate that multiple naphthalene moieties aggregate through π-stacking interactions.
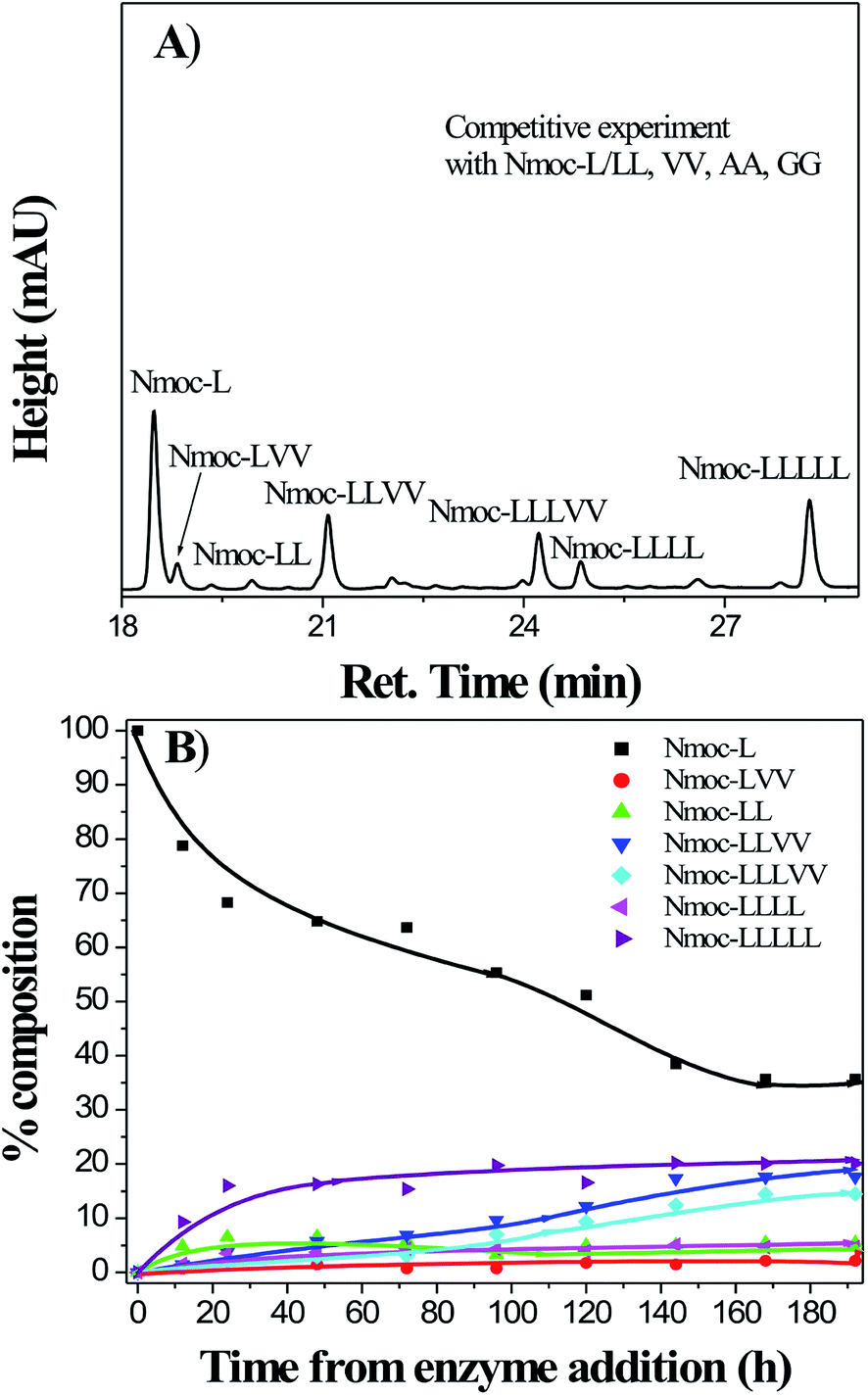
Time resolved fluorescence spectra were recorded to investigate the higher order aggregated state of the fluorophore groups in the hydrogel system (Fig. 9). The fluorescence decay profile has been fitted to the equation given below
where
τi is the fluorescence lifetime of various fluorescent species and
ai is the pre-exponential factors. All curves have been fitted with a tetra exponential decay curve. The time resolved fluorometry allows very precise discrimination between fluorophore species in different environments by contributing individual emission at the same wavelength. The hydrogel of the Nmoc-F/FF and Nmoc-L/LL systems shows a fluorescence decay lifetime of 1.07 ns and 0.267 ns, respectively (Table S1
†). The excitation wavelength was set at 367 nm and the emission was monitored at 468 nm. The fluorescence decay lifetime of the solution was found to be lower than hydrogels
1 and
2, which indicates a higher order aggregated nanofibrillar network
58 in the hydrogel (Fig. S18
†). In the gel state, the peptides aggregate in a more compact fashion. The peptides assemble from a rigid framework into a higher order structure through hydrogen bonding and π-stacking interactions, which results in an increased lifetime in the hydrogel state.
 |
| | Fig. 9 Emission decay curves of hydrogels 1 and 2 (concentration 20 mmol L−1) monitored at 468 nm (λex = 367 nm). The decay curve was recorded after 24 h of enzyme reaction. (IRF: instrument response function). | |
We used an oscillating rheometer to quantify the mechanical property of the hydrogels. The rheological measurement was performed with dynamic frequency sweep at a constant strain of 0.05% (Fig. S19†). The rheological measurements demonstrate the viscoelastic properties of the hydrogel (Fig. 10). The value of the storage modulus, G′, exceeds the value of the loss modulus, G′′, by an order of magnitude, which confirms the rigid- and solid-like behaviour of the hydrogels.59 The Nmoc-F/FF hydrogel system had a higher G′ than G′′ value, which confirms the rigid-, solid-like hydrogel. However, the Nmoc-L/LL hydrogel system is slightly weaker than hydrogel 1. Hydrogel 1 responds to gel–sol transition upon mechanical shaking and again restores its gel form upon resting. To quantify a thixotropic60,61 property of hydrogel 1, a simple step strain experiment was performed at constant angular frequency, 10 rad s−1. A constant strain of 0.05% was applied to hydrogel 1. Then the strain was slowly increased from 0.05 to 100%. The strain was kept constant for 2 min and 30 s, which resulted in gel-sol transition. The strain was decreased from 100% to 0.05% and the solution started to recover its gelation property (G′ > G′′) within 1 min and 40 s. The storage modulus (G′) was significantly higher than the loss modulus (G′′). The gel strength was recovered at 65% of its original stiffness after removing the strain (Fig. 11). However, hydrogel 2 was unable to show a thixotropic property.
 |
| | Fig. 10 Dynamic frequency sweep of (A) hydrogel 1 and (B) hydrogel 2 indicates that the storage modulus (G′) is higher than the loss modulus (G′′), which confirms the rigidity of the hydrogel at a constant strain of 0.05%. | |
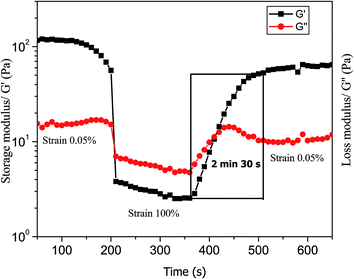 |
| | Fig. 11 Step strain experiment demonstrates the thixotropic property of hydrogel 1. A variation of storage modulus and loss modulus values was observed under the application and withdrawal of strain in a stepwise manner. | |
A microscopy study is used to reveal the formation of the nanostructures, which are responsible for the hydrogel formation. The hydrogels were analysed by transmission electron microscopy (TEM) and atomic force microscopy (AFM). Hydrogel 1 shows nanofibrillar networks, which is confirmed by TEM (Fig. 12A) and AFM (Fig. 12B). A preferred gelator component in a DCL forms 3D nanofibrillar networks, which is capable to entrap large volumes of water. TEM analysis revealed that the Nmoc-F/FF system self-assembled to form nanofibers62 with a diameter ranging from 35 to 45 nm (Fig. 12A). AFM analysis revealed a helical nanofibrillar morphology for hydrogel 1, with a height ranging from 8 to 12 nm (Fig. 12B). AFM analysis shows a nanofibrillar morphology (Fig. 13) for the Nmoc-L/LL system. The average height of the hydrogel 2 nanofibers is 25 nm. The existence of an interwoven morphology (Fig. S20†) indicates the formation of a self-supporting hydrogel.
 |
| | Fig. 12 (A) TEM image of hydrogel 1 shows a nanofibrillar morphology. (B) AFM image of hydrogel 1 indicates helical nanofibers. | |
 |
| | Fig. 13 AFM image of hydrogel 2 shows a nanofibrillar morphology. | |
Conclusion
Here, we describe an enzyme catalysed reverse hydrolysis reaction, which directs the peptide self-assembly in an aqueous medium. The self-assembly leads to the amplification of a preferred library component over the non-assembling components. The peptides, Nmoc-F3 and Nmoc-L5, evolved as major components for the Nmoc-F/FF and Nmoc-L/LL systems. The Nmoc-L/LLL system also depicts the formation of a preferred component, Nmoc-L5. The mechanical strength of the hydrogels was determined by rheological measurements, which indicate that both the hydrogels are strong and rigid. Hydrogen bonding as well as π-stacking interactions play major roles in the self-assembly process. The Nmoc-F/FF and Nmoc-L/LL systems form fibrous networks, which are responsible for the formation of a self-supporting hydrogel.
Experimental
Gel preparation
20 mmol of Nmoc-amino acid (Phe: F, Leu: L, Val: V and Ala: A) and 60 mmol of corresponding dipeptide and tripeptide (FF, LL, VV, AA and LLL) was suspended in 2 mL of 100 mmol sodium phosphate buffer (pH 8) in a glass vial. 1 mg mL−1 lyophilised thermolysin powder (bacillus thermoproteolyticus rokko from Sigma-Aldrich) was added to the reaction mixture. The mixture was vortex mixed for 15 s to ensure dissolution. A self-supporting hydrogel was formed after 5 min for the Nmoc-F/FF system, whereas the hydrogel formed after 36 h for the Nmoc-L/LL system.
HPLC analysis
A Dionex HPLC-Ultimate 3000 (High Performance Liquid Chromatography) pump was used to analyse the products. 20 μL of sample was injected onto a Dionex Acclaim ® 120 C18 column of 250 mm length with an internal diameter of 4.6 mm and 5 μm fused silica particles at a flow rate of 1 mL min−1 (linear gradient of 40% v/v acetonitrile in water for 35 min, gradually rising to 100% (v/v) acetonitrile in water at 35 min). This concentration was kept constant until 40 min when the gradient was decreased to 40% (v/v) acetonitrile in water at 42 min. The sample preparation involved mixing of 100 μL gel in 900 μL acetonitrile–water (50:50 mixture) solution containing 0.1% trifluoroacetic acid. The samples were then filtered through a 0.45 μm syringe filter (Whatman, 150 units, 13 mm diameter, 2.7 mm pore size) prior to injection. The products were identified by using an Ultimate 3000 RS Variable Wavelength Detector at 280 nm.
UV-Vis spectroscopy
UV-Vis absorption spectra of hydrogels 1 and 2 were recorded using a Varian Cary100 Bio UV-Vis spectrophotometer at a concentration of 20 mmol L−1.
Fluorescence spectroscopy
Fluorescence emission spectra of the gel (20 mmol L−1) were recorded from 325–620 nm with an excitation wavelength at 315 nm with medium sensitivity on a Horiba Scientific Fluoromax-4 spectrophotometer. The slit width for the excitation and emission was set at 2 nm and a 1 nm data pitch. The samples were prepared in a 1 cm2 quartz cuvette at room temperature.
Circular dichroism
Circular dichroism (CD) spectra were measured at 25 °C on a Jasco J-815 spectropolarimeter. The spectra were measured between 300 and 190 nm with a data pitch of 0.1 nm. The bandwidth was set to 1 nm with a scanning speed of 20 nm min−1 and a response time of 1 s. The path length was 1 mm quartz cell. The samples were prepared at a concentration of 2 mmol L−1. The experimental data were acquired in triplicate and the average data is shown. The CD data were obtained with the HT voltage less than 700 V.
FTIR
FTIR spectra were recorded using a Bruker (Tensor 27) FT-IR spectrophotometer with spectral resolution of 4 cm−1. All the measurements of the gel samples were performed on crystal Zn–Se windows and scanned between 900 and 4000 cm−1 over 64 scans.
Rheology
The rheological study was performed to determine the mechanical properties of the hydrogels. These properties were assessed using an Anton Paar Physica Rheometer (MCR 301, Austria) with cone plate geometry (20 mm in diameter, 50 μm gap and 1° angle) and the temperature was controlled at 25 °C. The dynamic moduli of the hydrogel were measured as a function of frequency in the range of 0.1–100 rad s−1 with a constant strain value of 0.05%. To determine the exact strain for the frequency sweep experiments, the linear viscoelastic (LVE) regime was performed at a constant frequency of 10 rad s−1. The experimental data were acquired in triplicate and the average data are shown. 200 μL of the gel was prepared in a glass vial and transferred over the plate using microspatula to proceed for the rheological measurements.
Morphological study
Transmission electron microscopy images were taken using a PHILIPS electron microscope (Model: CM 200), operated at an accelerating voltage of 200 kV. A dilute solution of the gel was dried on carbon-coated copper grids (300 mesh) by slow evaporation in air and then allowed to dry separately in vacuum at room temperature.
The morphology of the gels was investigated using tapping mode atomic force microscopy (AFM). The AFM study was done by placing a very dilute solution of the gel (200 μL of the gel was dissolved in 800 μL of Milli-Q water) on mica and allowed to dry in air for 2 days at room temperature. Images were recorded by using a scanning probe microscopy AIST-NT instrument (Model: Smart SPM-1000).
Time resolved study
2 mL gel was prepared in a 1 cm2 quartz cuvette and the time resolved studies were done by a time correlated single photon counting (TCSPC) system from Horiba Yovin (Model: Fluorocube-01-NL). The samples were excited at 376 nm using a picosecond diode laser (Model: Pico Brite-375L). The signals were collected at magic angle (54.70) polarization using a photomultiplier tube (TBX-07C) as detector, which has dark counts less than 20 cps. The instrument response function was typically 140 ps. The data analysis was performed using IBH DAS (version 6, HORIBA Scientific, Edison, NJ) decay analysis software.
The amplitude-weighted lifetime was estimated by
where
τ1 is the fluorescence lifetime of various fluorescent species and
a1 are the normalized pre-exponential factors. To gain the best fitting in all cases,
χ2 was kept near to unity.
Acknowledgements
AKD thanks CSIR for funding this work. DBR and MK are indebted to CSIR and SB is indebted to UGC for their fellowship. We thank SAIF, IIT Bombay for the assistance in the EM facility.
References
- H. C. Fry, J. M. Grarcia, M. J. Medina, U. M. Ricoy, D. J. Gosztola, M. P. Nikiforov, L. C. Palmer and S. I. Stupp, J. Am. Chem. Soc., 2012, 134, 14646–14649 CrossRef CAS PubMed.
- Y. Raz, B. Rubinov, M. Matmor, H. Rapaport, G. Ashkenasy and Y. Miller, Chem. Commun., 2013, 49, 6561–6563 RSC.
- D. M. Ryan and B. L. Nilsson, Polym. Chem., 2012, 3, 18–33 RSC.
- J. Y. Lee, Y. M. Kang, E. Kim, M. L. Kang, B. Lee, J. H. Kim, B. H. Min, K. Park and M. S. Kim, J. Mater. Chem., 2010, 20, 3265–3271 RSC.
- Z. Yang, G. Liang, M. Ma, A. S. Abbah, W. W. Lu and B. Xu, Chem. Commun., 2007, 843–845 RSC.
- D. Ma, K. Tu and L. M. Zhang, Biomacromolecules, 2010, 11, 2204–2212 CrossRef CAS PubMed.
- J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- H. Hosseinkhani, P.-D. Hong and D.-S. Yu, Chem. Rev., 2013, 113, 4837–4861 CrossRef CAS PubMed.
- C. A. E. Hauser and S. Zhang, Chem. Soc. Rev., 2010, 39, 2780–2790 RSC.
- L. Haines-Butterick, K. Rajagopal, M. Branco, D. Salick, R. Rughani, M. Pilarz, M. S. Lamm, D. J. Pochan and J. P. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7791–7796 CrossRef CAS PubMed.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1217 CrossRef CAS PubMed.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- S. Kiyonaka, K. Sugiyasu, S. Shinkai and I. Hamachi, J. Am. Chem. Soc., 2002, 124, 10954–10955 CrossRef CAS PubMed.
- M. D. Segarra-Maset, V. J. Nebot, J. F. Miravet and B. Escuder, Chem. Soc. Rev., 2013, 42, 7086–7098 RSC.
- I. Maity, D. B. Rasale and A. K. Das, Soft Matter, 2012, 8, 5301–5308 RSC.
- F. Versluis, H. R. Marsden and A. Kros, Chem. Soc. Rev., 2010, 39, 3434–3444 RSC.
- Z. Yang, G. Liang, L. Wang and B. Xu, J. Am. Chem. Soc., 2006, 128, 3038–3043 CrossRef CAS PubMed.
- J. Gao, H. M. Wang, L. Wang, J. Y. Wang, D. L. Kong and Z. M. Yang, J. Am. Chem. Soc., 2009, 131, 11286–11287 CrossRef CAS PubMed.
- J. H. Collier and P. B. Messersmith, Bioconjugate Chem., 2003, 14, 748–755 CrossRef CAS PubMed.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed.
- S. Ladame, Org. Biomol. Chem., 2008, 6, 219–226 CAS.
- J. M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- P. Vongvilai, M. Angelin, R. Larsson and O. Ramstrom, Angew. Chem., Int. Ed., 2007, 46, 948–950 CrossRef CAS PubMed.
- S. M. Turega, C. Lorenz, J. W. Sadownik and D. Philp, Chem. Commun., 2008, 4076–4078 RSC.
- N. A. Stephenson, J. Zhu, S. H. Gellman and S. S. Stahl, J. Am. Chem. Soc., 2009, 131, 10003–10008 CrossRef CAS PubMed.
- H. J. Cooper, M. A. Case, G. L. McLendon and A. G. Marshall, J. Am. Chem. Soc., 2003, 125, 5331–5339 CrossRef CAS PubMed.
- R. T. S. Lam, A. Belenguer, S. L. Roberts, C. Naumann, T. Jarrosson, S. Otto and J. K. M. Sanders, Science, 2005, 308, 667–669 CrossRef CAS PubMed.
- P. J. Boul, P. Reutenauer and J. M. Lehn, Org. Lett., 2005, 7, 15–18 CrossRef CAS PubMed.
- R. J. Lins, S. L. Flitsch, N. J. Turner, E. Irving and S. A. Brown, Angew. Chem., Int. Ed., 2002, 41, 3405–3407 CrossRef CAS.
- J. Hu, G. Zhang and S. Liu, Chem. Soc. Rev., 2012, 41, 5933–5949 RSC.
- S. Toledano, R. J. Williams, V. Jayawarna and R. V. Ulijn, J. Am. Chem. Soc., 2006, 128, 1070–1071 CrossRef CAS PubMed.
- R. J. Williams, A. M. Smith, R. Collins, N. Hodson, A. K. Das and R. V. Ulijn, Nat. Nanotechnol., 2009, 4, 19–24 CrossRef CAS PubMed.
- S. K. M. Nalluri and R. V. Ulijn, Chem. Sci., 2013, 4, 3699–3705 RSC.
- L. Chronopoulou, S. Lorenzoni, G. Masci, M. Dentini, A. R. Togna, G. Togna, F. Bordic and C. Palocci, Soft Matter, 2010, 6, 2525–2532 RSC.
- L. Chronopoulou, A. R. Togna, G. Guarguaglini, G. Masci, F. Giammaruco, G. I. Togna and C. Palocci, Soft Matter, 2012, 8, 5784–5790 RSC.
- M. G. Simpson, M. Pittelkow, S. P. Watson and J. K. M. Sanders, Org. Biomol. Chem., 2010, 8, 1181–1187 CAS.
- J. M. A. Carnall, C. A. Waudby, A. M. Belenguer, M. C. A. Stuart, J. J.-P. Peyralans and S. Otto, Science, 2010, 327, 1502–1506 CrossRef CAS PubMed.
- N. Sreenivasachary and J. M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5938–5943 CrossRef CAS PubMed.
- R. Orbach, L. Adler-Abramovich, S. Zigerson, I. Mironi-Harpaz, D. Seliktar and E. Gazit, Biomacromolecules, 2009, 10, 2646–2651 CrossRef CAS PubMed.
- Y. Zhang, H. Gu, Z. Yang and B. Xu, J. Am. Chem. Soc., 2003, 125, 13680–13681 CrossRef CAS PubMed.
- D. B. Rasale, I. Maity and A. K. Das, RSC Adv., 2012, 2, 9791–9794 RSC.
- H. Wang, C. Yang, M. Tan, L. Wang, D. Kong and Z. Yang, Soft Matter, 2011, 7, 3897–3905 RSC.
- L. Chen, S. Revel, K. Morrisb and D. J. Adams, Chem. Commun., 2010, 46, 4267–4269 RSC.
- K. Elfrink, J. Ollesch, J. Sçhr, D. Willbold, D. Riesner and K. Gerwert, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10815–10819 CrossRef CAS PubMed.
- M. S. Lamm, K. Rajagopal, J. P. Schneider and D. J. Pochan, J. Am. Chem. Soc., 2005, 127, 16692–16700 CrossRef CAS PubMed.
- X. Yan, Y. Cui, Q. He, K. Wang and J. Li, Chem. Mater., 2008, 20, 1522–1526 CrossRef CAS.
- B. Kim, S. Choi, S. Han, K. Choi and Y. Lim, Chem. Commun., 2013, 49, 7617–7619 RSC.
- S. G. Tarasova, V. Gaponenkob, O. M. Z. Howard, Y. Chenc, J. J. Oppenheimc, M. A. Dybaa, S. Subramaniame, Y. Leeb, C. Michejdaa and N. I. Tarasova, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(24), 9798–9803 CrossRef PubMed.
- E. F. Banwell, E. S. Abelardo, D. J. Adams, M. A. Birchall, A. Corrigan, A. M. Donald, M. Kirkland, L. C. Serpell, M. F. Butler and D. N. Woolfson, Nat. Mater., 2009, 8, 596–600 CrossRef CAS PubMed.
- P. Moitra, K. Kumar, P. Kondaiah and S. Bhattacharya, Angew. Chem., Int. Ed., 2013, 52, 1–6 CrossRef.
- X. Yan, Y. Cui, Q. He, K. Wang and J. Li, Chem. Mater., 2008, 20, 1522–1526 CrossRef CAS.
- C.-S. Chen, T.-J. Ji, X.-D. Xu, X.-Z. Zhang and R.-X. Zhuo, Macromol. Rapid Commun., 2010, 31, 1903–1908 CrossRef CAS PubMed.
- Y. Lim, K. Moon and M. Lee, Angew. Chem., Int. Ed., 2009, 48, 1601–1605 CrossRef CAS PubMed.
- H. Cao, X. Zhu and M. Liu, Angew. Chem., Int. Ed., 2013, 52, 4122–4126 CrossRef CAS PubMed.
- Y. Kuang, Y. Gao, J. Shi, H. Lin and B. Xu, Chem. Commun., 2011, 47, 8772–8774 RSC.
- Z. Yang, L. Wang, J. Wang, P. Gao and B. Xu, J. Mater. Chem., 2010, 20, 2128–2132 RSC.
- A. K. Das, A. R. Hirst and R. V. Ulijn, Faraday Discuss., 2009, 143, 293–303 RSC.
- I. Maity, T. K. Mukherjee and A. K. Das, New J. Chem., 2014, 38, 376–385 RSC.
- S. Roy and A. Banerjee, Soft Matter, 2011, 7, 5300–5308 RSC.
- S. Basak, J. Nanda and A. Banerjee, Chem. Commun., 2014, 50, 2356–2359 RSC.
- Y. Ohsedo, M. Oono, K. Saruhashib and H. Watanabeab, RSC Adv., 2014, 4, 48554–48558 RSC.
- J. Li, Y. Gao, Y. Kuang, J. Shi, X. Du, J. Zhou, H. Wang, Z. Yang and B. Xu, J. Am. Chem. Soc., 2013, 135, 9907–9914 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra09490e |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.