
Kinetic Monte Carlo simulations of the assembly of filamentous biomacromolecules by the dimer addition mechanism†
 *a
and
*a
and
Abstract
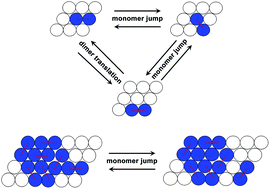
In cells, several important biomacromolecules form oligomers through a dimer addition mechanism. Rate equations based on mean field approximations are usually employed to describe the assembly process. However, such equations often require multiple assumptions that mask some detailed changes of the biomolecular configurations during aggregations. Here, we present a kinetic Monte Carlo simulation scheme to account for the diffusion and rotation of dimers on two-dimensional hexagonal lattices while naturally including the stochastic features. We investigate the effects of the interaction energy between dimers, the diffusion coefficient and the concentration of dimers on the aggregation by dimer addition mechanism. Our simulations identified unusual double-S shape evolutions of aggregation kinetics, which are probably associated with the formation of metastable clusters.
 Please wait while we load your content...
Please wait while we load your content...

