
I2 catalyzed tandem protocol for synthesis of quinoxalines via sp3, sp2 and sp C–H functionalization†
Abstract
One-pot, atom-economic synthesis of quinoxalines has been achieved through generation of arylglyoxal from easily available ethylarenes, ethylenearenes and ethynearenes, and subsequent condensation with o-phenylenediamines. Catalytic I2 with TBHP as an oxidant in DMSO is the system of choice for this domino reaction involving C–H functionalization/oxidative cyclization. This metal-free, mechanistically distinct and functional group tolerant tandem approach could be a powerful complement to traditional approaches for the synthesis of quinoxalines.
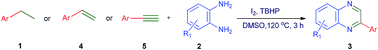
 Please wait while we load your content...
Please wait while we load your content...

