
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRhodium catalysed conversion of carbenes into ketenes and ketene imines using PNN pincer complexes†
Z.
Tang
a,
S.
Mandal
a,
N. D.
Paul
a,
M.
Lutz
b,
P.
Li
a,
J. I.
van der Vlugt
a and
B.
de Bruin
*a
aSupramolecular, Homogeneous & Bio-inspired Catalysis, van't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, The Netherlands. E-mail: b.debruin@uva.nl
bCrystal and Structural Chemistry, Bijvoet Center for Biomolecular Research, Utrecht University, Padualaan 8, 3584 CH, Utrecht, The Netherlands
First published on 15th October 2015
Abstract
Ketene synthesis involving catalytic carbonylation of carbenes is an interesting alternative to traditional synthetic protocols, offering milder conditions to diversified products. Analogous catalytic ketene imine production from carbenes and isocyanides is also a promising reaction. However, both methods are underdeveloped. Rhodium carbonyl complexes B and E, based on (6-(phosphinomethyl)pyridin-2-yl)methan-sec-amine type PNN ligand scaffolds, reveal good catalytic activities in ketene and ketene imine production using ethyl diazoacetate (EDA, 1) or sodium 2-benzylidene-1-tosylhydrazin-1-ide (5) as the carbene precursors, as demonstrated by in situ amide/imidamide and β-lactam synthesis. DFT calculations suggest that diazo activation is the rate-determining step and that NH-deprotonation of the ligand produces a more active rhodium complex. The ketene formation step likely proceeds via an outer-sphere CO insertion mechanism. Subsequent stepwise and concerted [2 + 2] cyclization mechanisms have comparable barriers. The complexes are the first rhodium catalysts reported for catalytic ketene/ketene imine production from carbenoids. The higher affinity of rhodium for binding ketene or ketene imine intermediates as compared to other reported metal catalysts (i.e. Pd, Co) may provide opportunities for future enantioselective reactions when using chiral ligands.
Introduction
Ketenes1 are highly reactive and useful intermediates to a wide range of important chemicals for pharmaceuticals and fine chemicals, such as esters, amides, β-lactons or β-lactams.2 Ketene imines, the isoelectronic nitrogen analogues, are also reactive and versatile intermediates that tend to undergo analogous coupling reactions to give relevant nitrogen-containing compounds.2b,3Ketenes are conventionally synthesized by thermolysis,4 Wolff rearrangement5 or dehydrohalogenation of acyl halide precursors.6 These methods generally have limitations, such as a narrow substrate scope, strictly controlled conditions and generation of detrimental side products. Ketene imines are generally synthesized from ketenes via an aza-Wittig reaction,7 various rearrangement reactions8 or azide–alkyne coupling.9 These methods have similar limitations concerning the substrate scope and reaction conditions.
Ketene chemistry has a close connection with carbenes.10 Carbonylation of carbenes to give ketene formation was reported in 1912,11 and the reactivity to undergo carbonylation has remained a characteristic feature of carbenes. A number of early or late transition metal complexes have been reported to mediate carbonylation of carbenes to form ketenes.10,12 This strategy to construct ketenes should allow a larger substrate scope, as a wide variety of diazo-compounds and their precursors are easily accessible. Possibly, also milder reaction conditions should be achievable. However, most reported reactions are strictly stoichiometric and usually require a high CO pressure and/or high reaction temperatures.
The catalytic version of ketene synthesis from carbonylation of carbenes has been largely underdeveloped,13,14 with cobalt14b,d,f and palladium14e catalysts being reported only very recently. Ketenes were generated with these catalysts in decent yields under relatively mild conditions (1–20 bar of CO, 50–60 °C), and in situ converted for the synthesis of a range of esters, amides and β-lactams (Scheme 1), using diazo14 and N-tosylhydrazone15 compounds as carbene precursors.
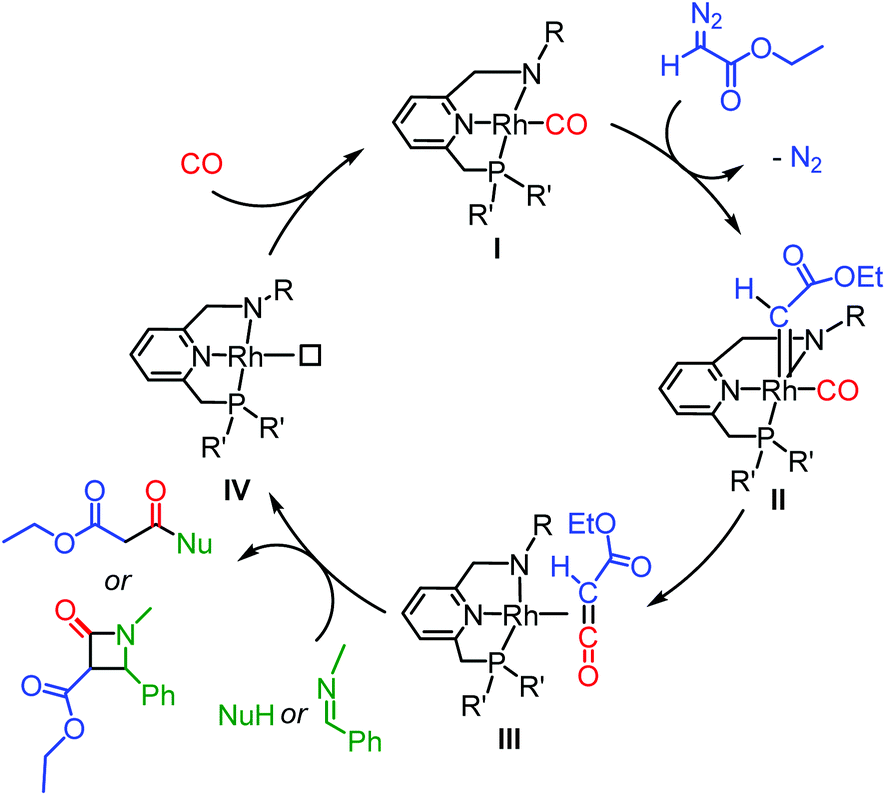 | ||
| Scheme 1 The envisioned catalytic cycle of ketene formation and subsequent coupling reaction mediated by Rh PNN complexes (only inner-sphere CO insertion pathway depicted). | ||
Although stereocontrol in the coupling reactions of ketene is highly desirable,1,16 the transition metal catalysts reported thus far for ketene formation from carbene precursors are not able to promote the follow-up coupling reactions in an enantioselective or diastereoselective manner (e.g. when several chiral Co(porphyrin) complexes were used as catalysts, no ee was detected14f). This is attributed to a low affinity of the metal ions to bind the ketene intermediate, which as a result undergoes coupling reactions after its release from the metal centre.
A single molecular catalyst that can catalyse ketene formation from carbenes and can induce stereoselectivity in the coupling reactions would be highly desirable. Our consideration on catalyst development for this purpose was to utilize the Rh(PNN) scaffold developed earlier in our lab, as rhodium has been recognized as one of the most active metals to mediate the carbonylation of carbenes to form ketenes,10a and rhodium should have a relatively high affinity to bind the ketene intermediates, of key-importance for future chirality transfer. In addition, the PNN ligand is a versatile scaffold with both the amine and the phosphine available to install chiral auxiliaries in future studies. The rigidity of the pincer ligand is also expected to provide stability to the metal complex during the reaction. Furthermore, derivatives of the complexes upon –NH deprotonation are accessible, offering easy modification and diversity in the number of complexes to screen for catalytic activity. While the studies in this report focus on achiral model systems, the results are nonetheless relevant for future development of enantioselective catalysis.
In the envisioned catalytic working model, the diazo compound (ethyl diazoacetate, EDA, is taken as an example in Scheme 1) first coordinates to Rh complex I before being converted to the metallocarbene intermediate IIvia N2 loss. Subsequent CO insertion into the metallocarbene (via either an inner- or outer-sphere mechanism; the inner-sphere mechanism being shown in Scheme 1) then results in formation of the metal-ketene species III. Follow-up reactions, such as nucleophilic coupling or [2 + 2] imine-ketene cyclization, can occur in the coordination sphere of the metal. If the PNN ligand bears chiral substituents, enantioselective coupling may be envisioned. After the coupling, release of the product and (possible) coordination of exogenous CO would close the catalytic cycle. Ketene imines can be prepared in a similar manner by using isocyanides in place of CO.3 Examples of the stoichiometric reaction are not abundant,17 but examples of catalytic synthesis of ketene imines are even scarcer and limited to only two recently reported palladium catalysts.18 We therefore considered it worthwhile to develop a protocol for catalytic metal-mediated ketene imine synthesis, and these studies are included herein.
The main goals of the investigations presented here are: (I) To show that Rh(PNN) complexes are viable carbene-transfer catalysts for ketene and ketene imine synthesis, and (II) to investigate computationally whether the overall catalytic reaction could proceed via rhodium-coordinated ketenoid intermediates that undergo attack by external nucleophiles in the coordination sphere of rhodium (the latter being obviously relevant for future development of enantioselective strategies with chiral analogs of the complexes studied herein).
Results and discussion
Synthesis of the PNN ligands
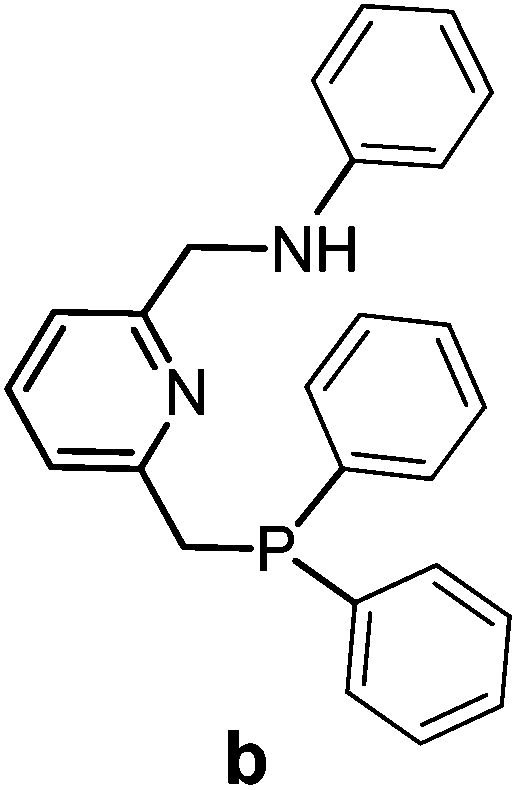
The PNN ligands a and b, with phenyl substituents at phosphorus, were synthesized via pathway [1] (Scheme 2), a modification from a previously reported procedure.19 Synthesis of a and b starts with condensation of 6-methylpicolinaldehyde with the desired amines, followed by imine reduction to the amines. The isolated amines were treated with two equivalents of n-butyl lithium (nBuLi) in diethyl ether (Et2O) at −78 °C, with subsequent cannulation of this reaction mixture to a solution of Ph2PCl in Et2O at −78 °C and acidic work-up to afford the PNN ligands a and b in 15–20% overall yields.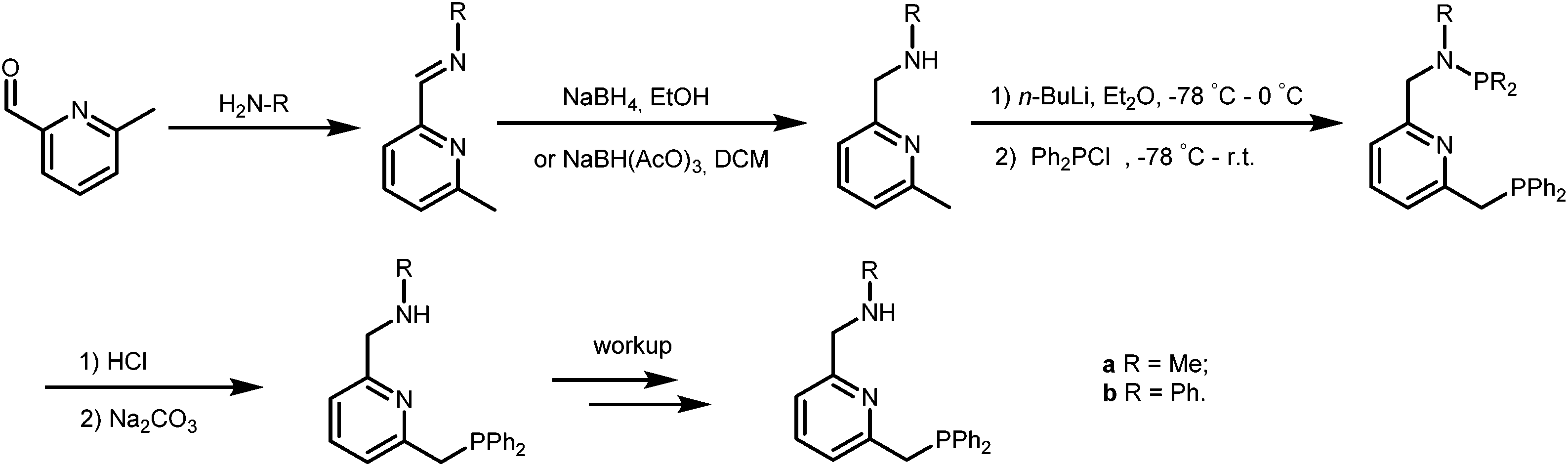 | ||
| Scheme 2 Synthetic pathway [1] for the synthesis of ligands a and b. | ||
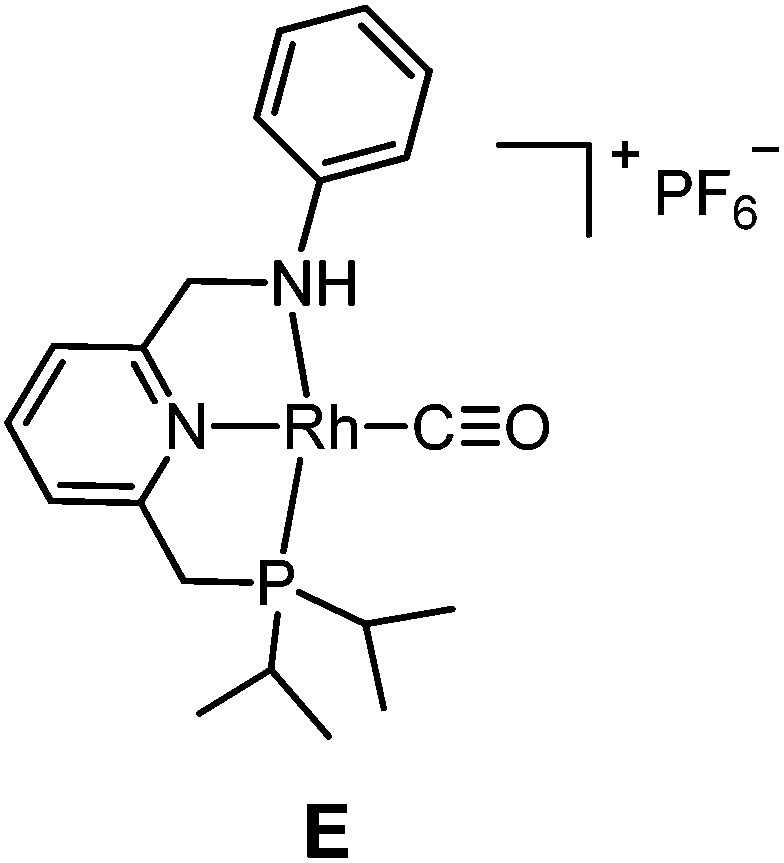
The PNN ligands c–e, which bear alkyl substituents at phosphorus, were synthesized via pathway [2] (Scheme 3), also following a modified literature procedure.20 Borane was used as protecting group for the alkylphosphine intermediates to allow purification processes to be performed under aerobic conditions. Subsequent installation of the phosphine arm and the nitrogen arm on 2,6-bis(chloromethyl)pyridine followed by borane removal afforded the pincer ligands c, d and e in 34–44% overall yields.
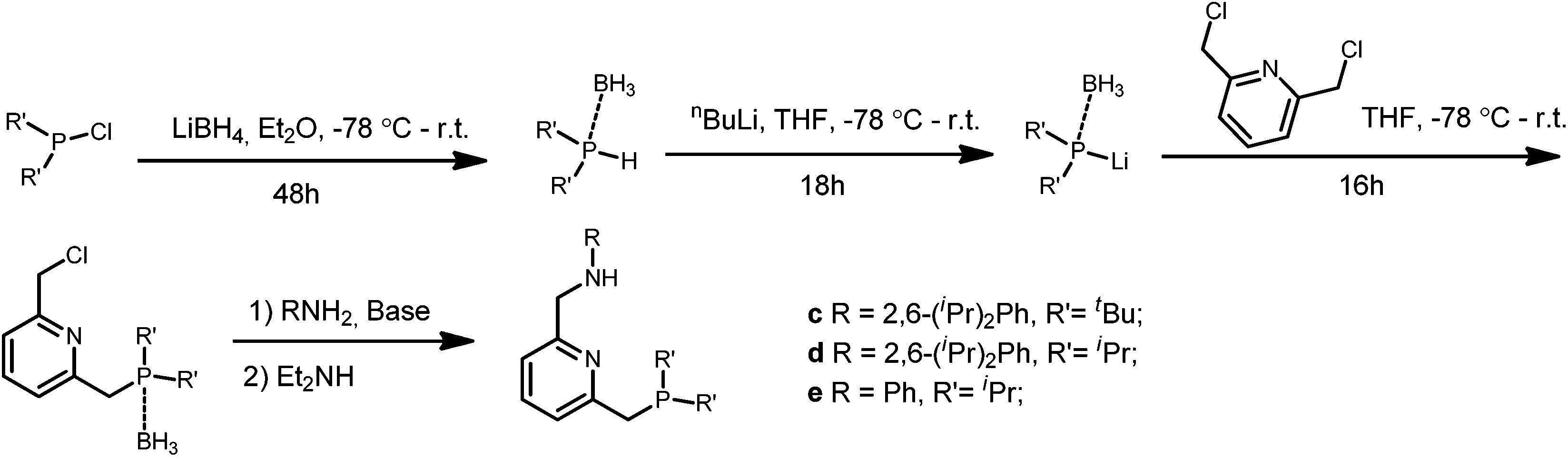 | ||
| Scheme 3 Synthetic pathway [2] for the synthesis of ligands c–e. | ||
Synthesis of the PNN rhodium carbonyl complexes
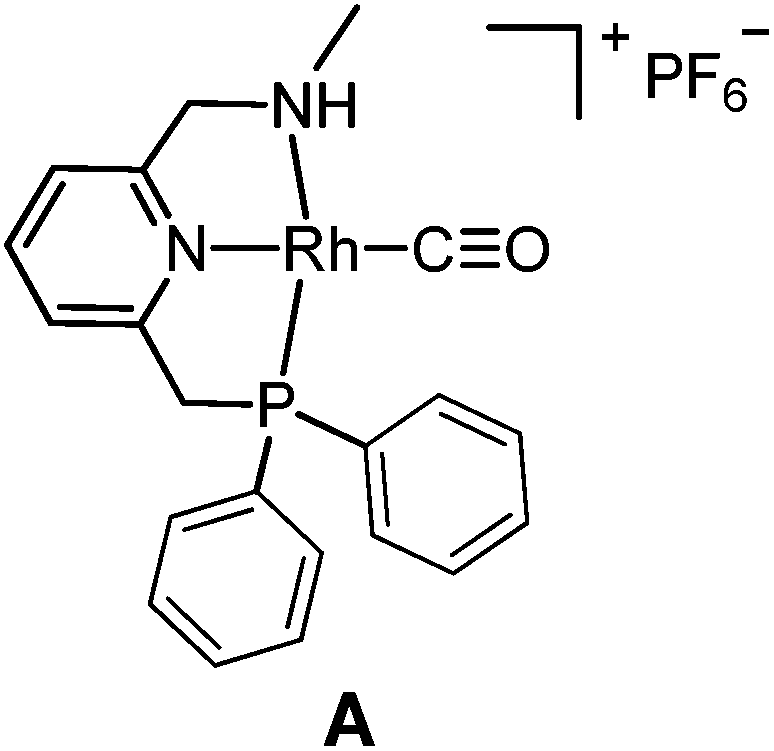
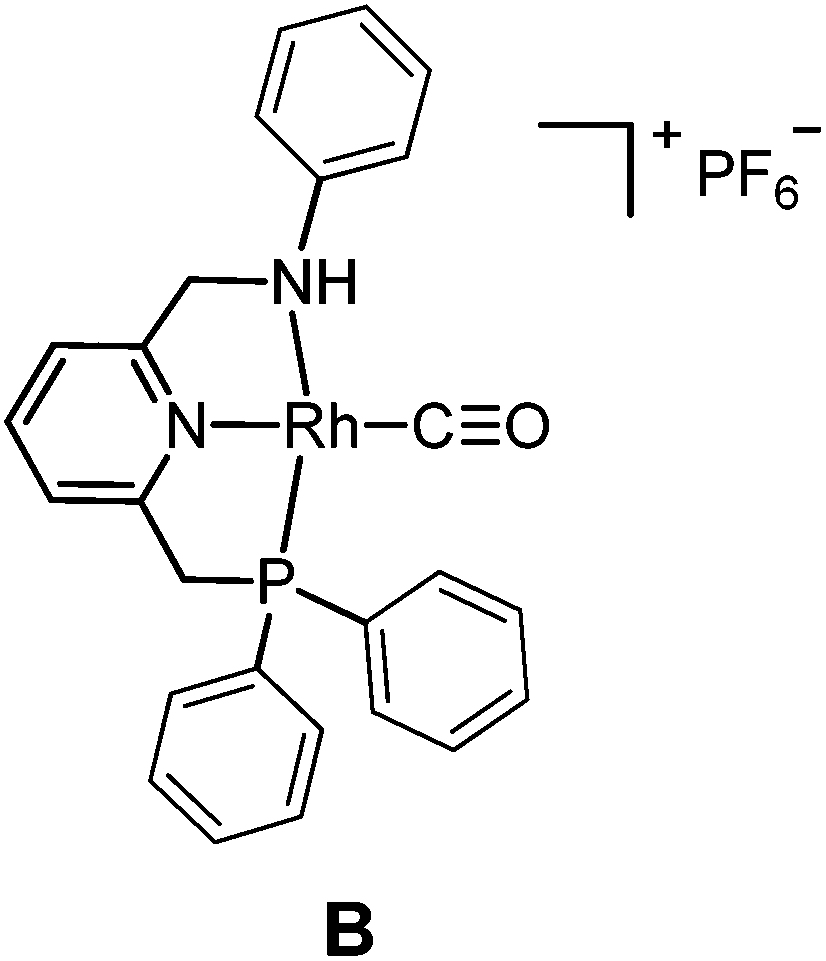
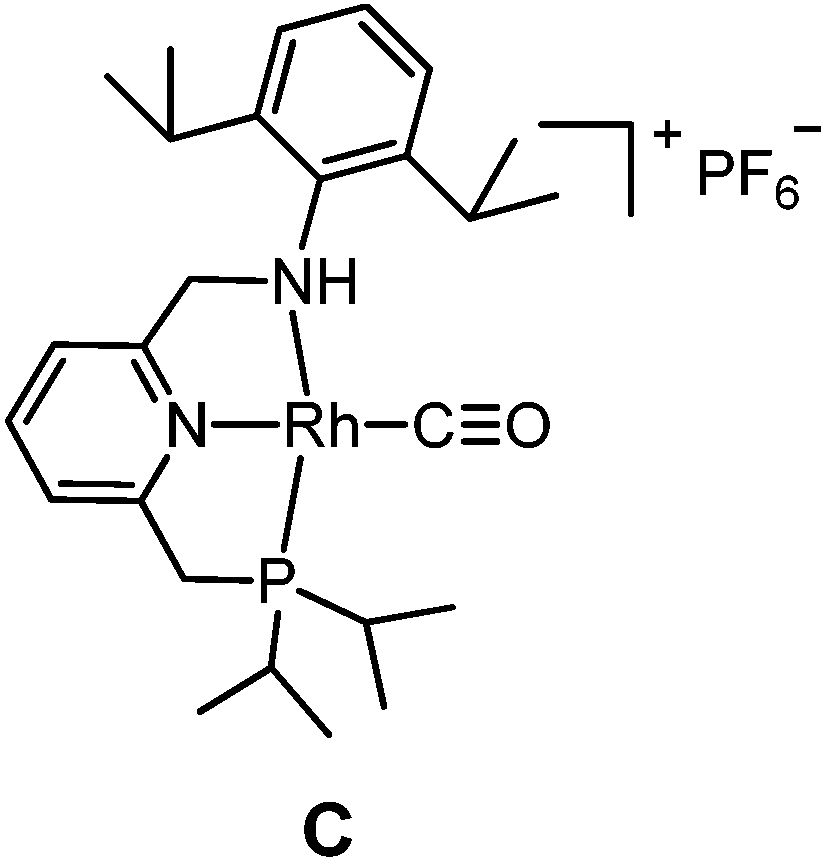
The targeted Rh(CO)(PNN) complexes were obtained by reaction of {[Rh(CO)2(μ-Cl]2} with two equivalents of the respective PNN ligands in methanol, followed by salt metathesis using NH4PF6 (Scheme 4).21 Five complexes were synthesized and isolated. Complexes A and B both have a phenyl-substituted phosphine arm, only differing in the substituent on the nitrogen arm (methyl for A and phenyl for B). Complexes C and D both have bulky 2,6-isopropylphenyl substituents on the nitrogen arm. They differ in the substituents at phosphorus atom (isopropyl for C and tert-butyl for D). Complex E is an analogue of complex D with a phenyl substituent on the nitrogen arm. Deprotonation of the amine moieties of cationic complexes B–E produces the neutral amido complexes B′–E′ (Scheme 4),22 but these were not isolated.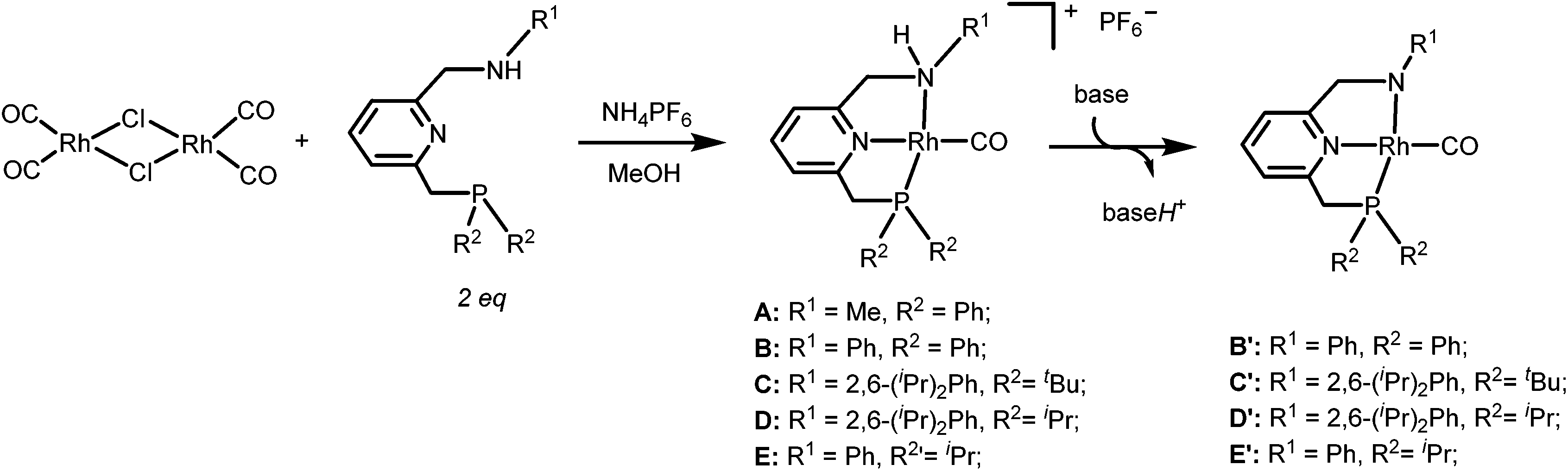 | ||
| Scheme 4 Synthesis of cationic amine Rh(CO)(PNN) complexes A–E and their neutral amido analogues A′–E′. | ||
Yellow single crystals of complex A were obtained by storing a concentrated methanol solution of A at 0 °C, while crystalline material for B and E was obtained by top-layering the corresponding concentrated dichloromethane (DCM) solution with diethyl ether (Et2O). The X-ray crystal structures23 of the three complexes are depicted in Fig. 1, and selected bond lengths and angles are listed in Table 1. All the three complexes adopt a slightly distorted square planar geometry around Rh, with the plane defined by the atoms P1, N1 and N2. Bond lengths and angles around Rh in these complexes are similar.
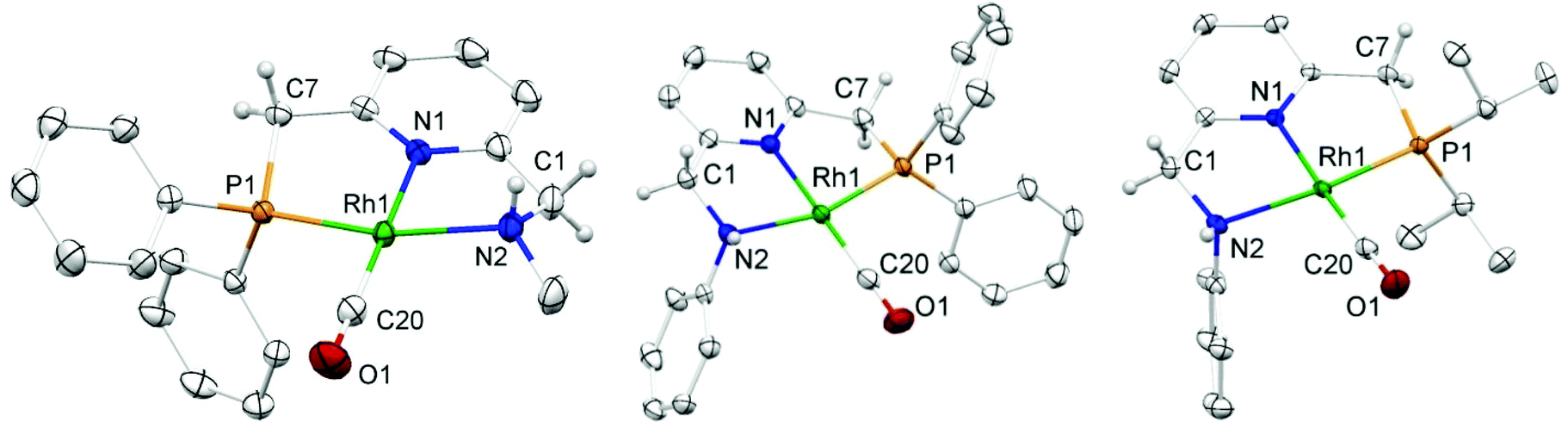 | ||
| Fig. 1 Molecular structures (ORTEP at 50% level) of the cationic complexes A (left), B (middle) and E (right). Counter ions and hydrogen atoms are removed for clarity, except for those on C1, C7 and N2. In A, only the major disorder form of the coordinated amine group is shown. | ||
| A | B | E | |
|---|---|---|---|
| Rh1–P1 | 2.2234(3) | 2.2120(9) | 2.2126(4) |
| Rh1–N1 | 2.0540(9) | 2.053(3) | 2.0483(12) |
| Rh1–N2 | 2.1381(10) | 2.160(3) | 2.1624(13) |
| Rh1–C20 | 1.8283(12) | 1.831(4) | 1.8383(16) |
| C20–O1 | 1.1489(15) | 1.147(5) | 1.139(2) |
| P1–Rh1–N1 | 82.83(2) | 83.59(8) | 84.77(3) |
| P1–Rh1–N2 | 161.78(3) | 163.53(9) | 161.18(4) |
| N1–Rh1–C20 | 176.57(4) | 175.19(15) | 176.44(6) |
| Rh1–C20–O1 | 178.01(12) | 176.0(4) | 178.64(17) |
Slightly longer Rh–N2 bonds in B and E than in A (2.1609 and 2.1626(13) vs. 2.1381(10) Å) reflect a slightly weaker donor capacity of the N2 donor in complexes B and E. The infrared spectra in CD2Cl2 solution show a higher CO stretching frequency in B than in A (2007 vs. 2002 cm−1) is in agreement with weaker metal-to-carbonyl back-donation in B. Lower CO stretching frequencies in complexes C, D and E were observed (1999, 1996 and 1999 cm−1, respectively), as these ligands bear more electron-rich alkylphosphine ligands.
Catalytic results
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | 2 eq. K2CO3 | 3a + 4 yield (%) | 3a/4 ratio | Remarks |
| 1 | A | × | 50 | 0.4 | |
| 2 | A | √ | 26 | 0.2 | |
| 3 | B | × | 100 | 0.4 | 38 h |
| 4 | B | √ | 100 | 7.4 | 38 h |
| 5 | B | √ | 94 | 9.7 | |
| 6 | C | √ | 12 | 0.6 | |
| 7 | D | √ | 14 | 0.4 | |
| 8 | E | √ | 100 | >50 | |
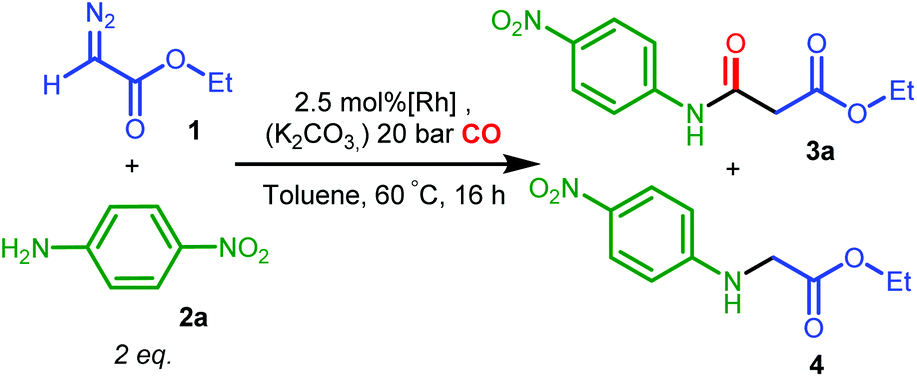
For complex A, poor selectivities with moderate or low yields to the desired amide product 3a were obtained, either in the absence or presence of K2CO3 (entries 1 and 2, Table 2). In contrast, the diphenylphosphine complex B with phenyl substituents on the amine nitrogen gave much better results in the presence of the base, both in terms of yield and selectivity to 3a (entries 4, 5, the small selectivity difference might originate from overlap of signals used for integration in the 1H NMR spectra), while amine 4 was obtained as the major product in the absence of the base.
The nucleophilic coupling with the ketene appears to be accelerated to a larger extent than the side reaction under these basic conditions. This suggests that amine deprotonation of B to form the neutral amido complex B′ (see Scheme 4) is beneficial for the selectivity.
One might hypothesize that for the neutral amido complex B′ the carbene intermediate II is less prone to external attack by the aniline 2a, due to increased π-back donation to the carbene p-orbital (as a result of increased electron density) as compared to the cationic amine complex B, whereas CO migration or external CO attack to the carbene moiety to form the ketene might be facilitated by the increased nucleophilicity of the carbene ligand (taking advantage of the electron-accepting properties of CO). Control experiments showed that complex A was inert toward deprotonation by K2CO3, while B was readily deprotonated by K2CO3 in THF,24 consistent with the assumption that a neutral amido intermediate is the active species.
The poor performance of complexes C and D is probably related to steric shielding by the bulky side-groups in these ligands, while the less bulky complex E (in its deprotonated form E′) gave almost full conversion and a very high selectivity (3a/4 ratio >50) to 3a (entry 8) in 16 hours. Deprotonated complex E′ clearly outperforms all other catalysts screened in this study, both in terms of activity and selectivity. Complexes B′ and (in particular) E′ are thus determined to be supreme catalysts for ketene generation with EDA as the substrate.
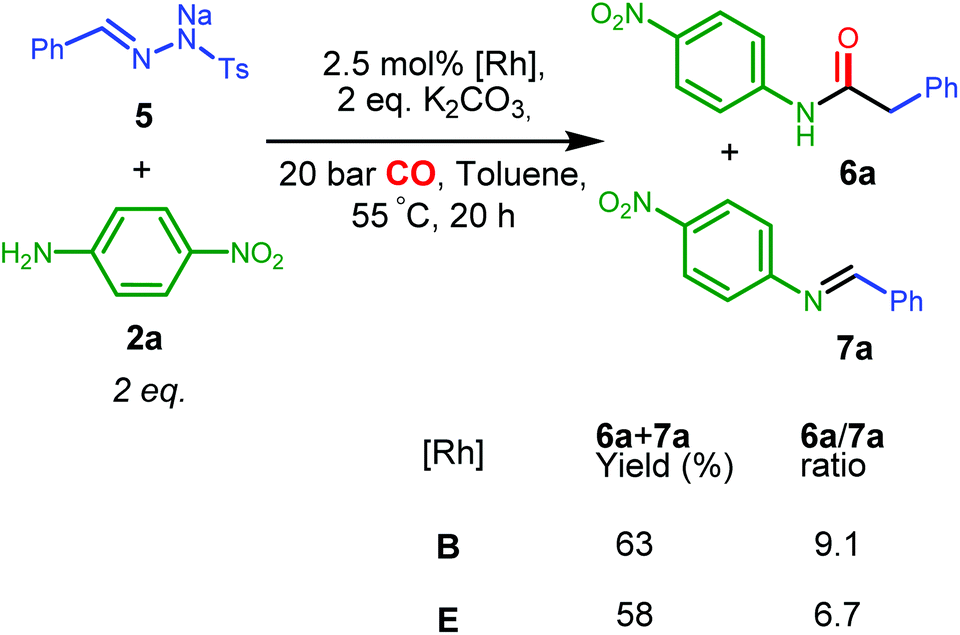 | ||
| Scheme 5 Catalytic activities of complexes B and E for synthesis of amide 6a from 5 and 2a. | ||
β-Lactam synthesis via catalytic ketene formation
β-Lactams are medicinally important structures and versatile building blocks for other valuable compounds.27 A number of methods are available to synthesize these compounds, with [2 + 2] Staudinger ketene-imine cyclization being among the most extensively used and studied protocols.28 Recently, novel synthetic protocols were developed that enhance the synthetic applicability of Staudinger-type β-lactam synthesis, making use of in situ generated ketenes via catalytic carbonylation of diazo-derived carbenes in one-pot synthetic protocols to prepare the respective β-lactams.14 This was shown to be a useful alternative to more traditional β-lactam synthesis. The next step in this field is to develop enantioselective versions of these reactions. However, this is not so trivial, as the ketene intermediates seem to have low affinity for the catalysts applied thus far. Hence we studied the Rh(PNN) complexes as catalysts for this reaction.Using similar conditions as the recently reported literature,14e,f we tested the catalytic activity of the complexes in the reaction using N-benzylidenemethanamine 8a as the imine substrate.
Surprisingly, when EDA was used as the substrate, the complexes B or E, identified as supreme catalysts in catalytic ketene synthesis for amide formation, gave generally no formation or less than 5% yield to the expected β-lactam product, either in the presence or absence of K2CO3, with no identifiable side products and in many cases unreacted EDA detected after the reaction. The exact reason behind this behaviour is not clear, as EDA can be used in catalytic ketene formation as demonstrated by the reactions described in Table 2. Nonetheless, EDA and imine 8a are poor coupling partners in catalytic formation of β-lactams when using catalyst B or E. The Rh complexes tested are thus not compatible with the carbene precursor EDA for the corresponding β-lactam synthesis under the currently applied conditions.
In marked contrast to the results obtained with EDA, using the N-tosylhydrazone salt 5 as the carbene precursor led to more selective reactions, producing the β-lactam product 9a (Table 3). In the series of complexes A–C and E, complex B gave the highest yield of the desired β-lactam product 9a (67.7%), while E (which was one of the best performing catalysts for the reactions described in Table 2) gave only a moderate yield of 33.0%. Complex A gave no formation of β-lactam, while complex C led to low yield and 23.1% of the (free) phenyl ketene, according to 1H NMR spectroscopy. It should be noted that 5 is also a relative strong base, hence the corresponding Rh complexes are expected to exist in their NH-deprotonated neutral amido forms in the catalytic reaction, even in the absence of additional base. In all cases, the trans-configured β-lactam was the predominant isomer, while the cis-isomer was either not detected or present as a minor product (assignments based on literature14e). The trans–cis stereoselectivity has been a poorly understood and controversial topic in the Staudinger reaction.29 Previous studies have shown that the stereoselectivity may be related to isomerization of the imine or zwitterionic intermediate, and is determined by many factors such as solvent and temperature, but most importantly the substituents of both the ketene and the imine. As higher trans/cis ratios of the product 9a were generally observed with the rhodium catalyst system than for non-catalysed systems29c and those with Pd14e and Co14f systems under similar or identical conditions, we believe that the cyclization process is also metal-catalysed.30 Metal-involvement in the [2 + 2] ketene-imine cyclization has been suggested for ketenes generated from carbonylation of Fischer carbenes.31 As such, this is a promising observation, stimulating further research in the development of enantioselective reactions.
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Yield of 9a (%) | Yield of 10 (%) |
| 1 | A | 0 | |
| 2 | B | 67.7 | |
| 3 | C | 7.2 | 23.1 |
| 4 | E | 33.0 | |
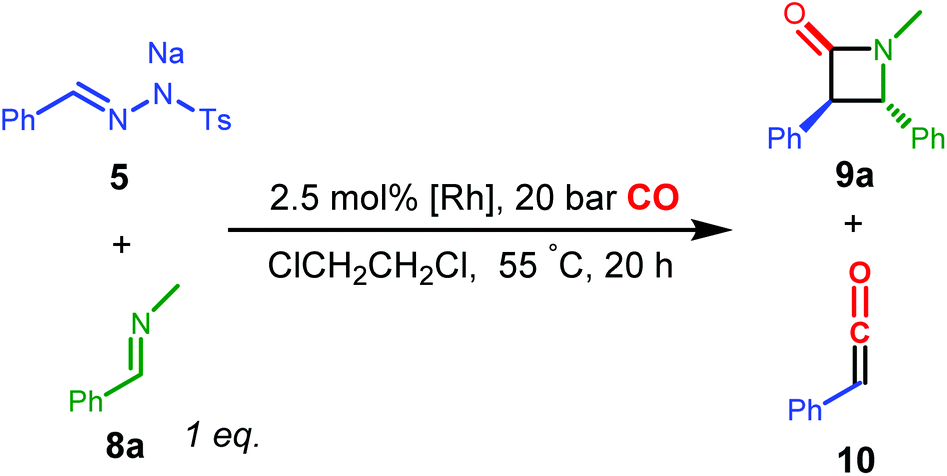
Variation of the para-substituent on the phenyl ring of imine 8 has an effect on the yield of products 9, but in all cases moderate to excellent activities for coupling with the in situ generated ketene were observed (see ESI, Table S2†).
Catalytic ketene imine synthesis from carbene precursor 5 and tert-butyl isocyanide 11
Ketene imines, the nitrogen analogues of ketenes (e.g.12 in Scheme 6), represent an important class of reactive species and useful synthetic intermediates, which are able to undergo nucleophilic coupling or cyclization reactions for the production of various pharmaceutically valuable molecules, e.g. substituted isoquinolines and pyridines.2b,3 As the rhodium complex B showed good catalytic activities in mediating ketene formation via carbonylation of carbenes, we were interested to extend the synthetic concept to catalytic ketene imine synthesis from carbenes and isocyanides, which are isoelectronic to CO. As this reaction has thus far mostly been studied stoichiometrically,17 development of catalytic methods should be practically useful,18 potentially allowing a wide substrate scope and an alternative mild condition route to traditional ketene imine synthesis.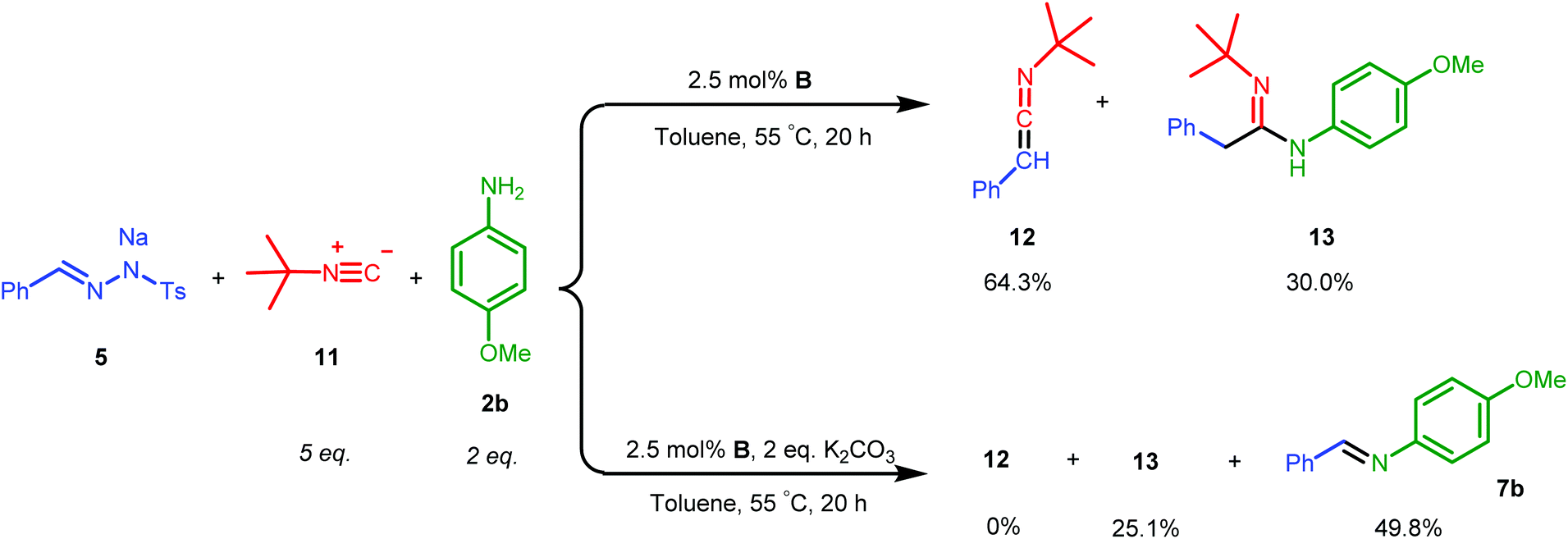 | ||
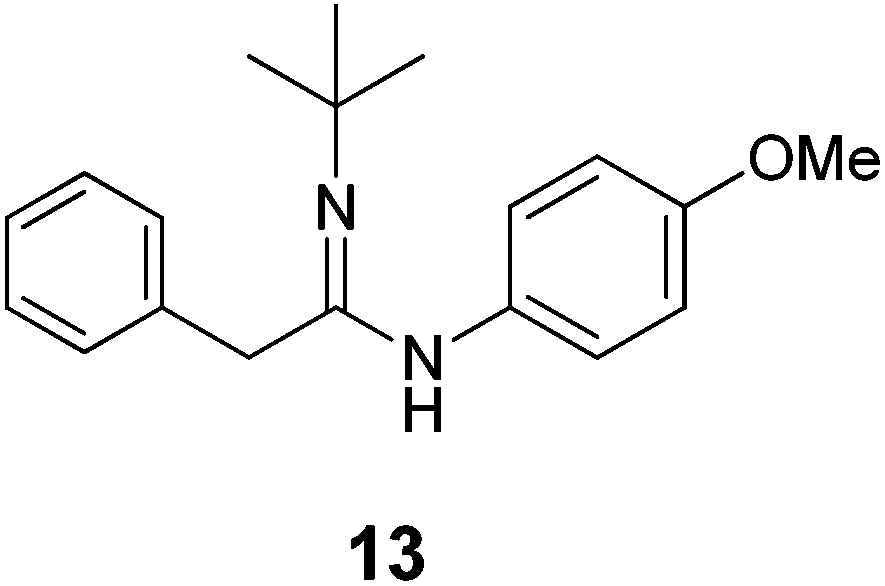
| Scheme 6 Acetimidamide 13 synthesized by one-pot catalytic ketene imine formation from 5 and 11 and subsequent trapping of ketene imine 12 by aniline 2b. | ||
While EDA turned out to be an unsuitable carbene precursor for the reaction (no ketene imine was observed after a series of reaction optimizations with and without K2CO3), the hydrazone salt 5 in combination with tert-butyl isocyanide 11 gave 50.7% yield of the phenyl ketene imine 12, catalysed by complex B at 55 °C overnight (see ESI, Table S3†). Excess 11 is beneficial, producing ketene imine 12 in a higher yield (entries 1 and 2, Table S3†). To the best of our knowledge, this is the first example of catalytic synthesis of any ketene imine from a carbene precursor and an isocyanide. Different batches of 5 again led to irreproducibility of yields and a need to use a higher reaction temperature to covert certain batches of tosyl hydrazone salt 5 to the diazo compound (see Table S3†).
We next investigated the one-pot synthesis of 2-phenylacetimidamide 13 by adding an amine nucleophile 2b to the in situ generated ketene imine (Scheme 6), which led to 30.0% yield of the desired product 13 along with 64.3% of the intermediate ketene imine 12, despite a reaction time of 44 hours. This possibly relates to the lower electrophilicity of ketene imines as compared to ketenes, or (perhaps more likely) serves as an indication that the coupling reaction is also catalysed by the metal complex, which is possibly slowed down by catalyst deactivation over the course of the reaction. Addition of two equivalents of K2CO3 was expected to accelerate the coupling process. However, while the ketene imine 12 was not observed after 18 hours, the 2-phenylacetimidamide 13 was obtained only in 25.1% yield, and the imine product 7b was obtained as the major product in 49.8% yield. Further optimizations by varying the base, solvent and ratio of agents could result in selectivity improvements, but this was not attempted because it appears right now that the Rh(PNN) systems are too sensitive to allow high conversions of 12 to 13 in one-pot transformations using the same catalyst.
Mechanistic studies
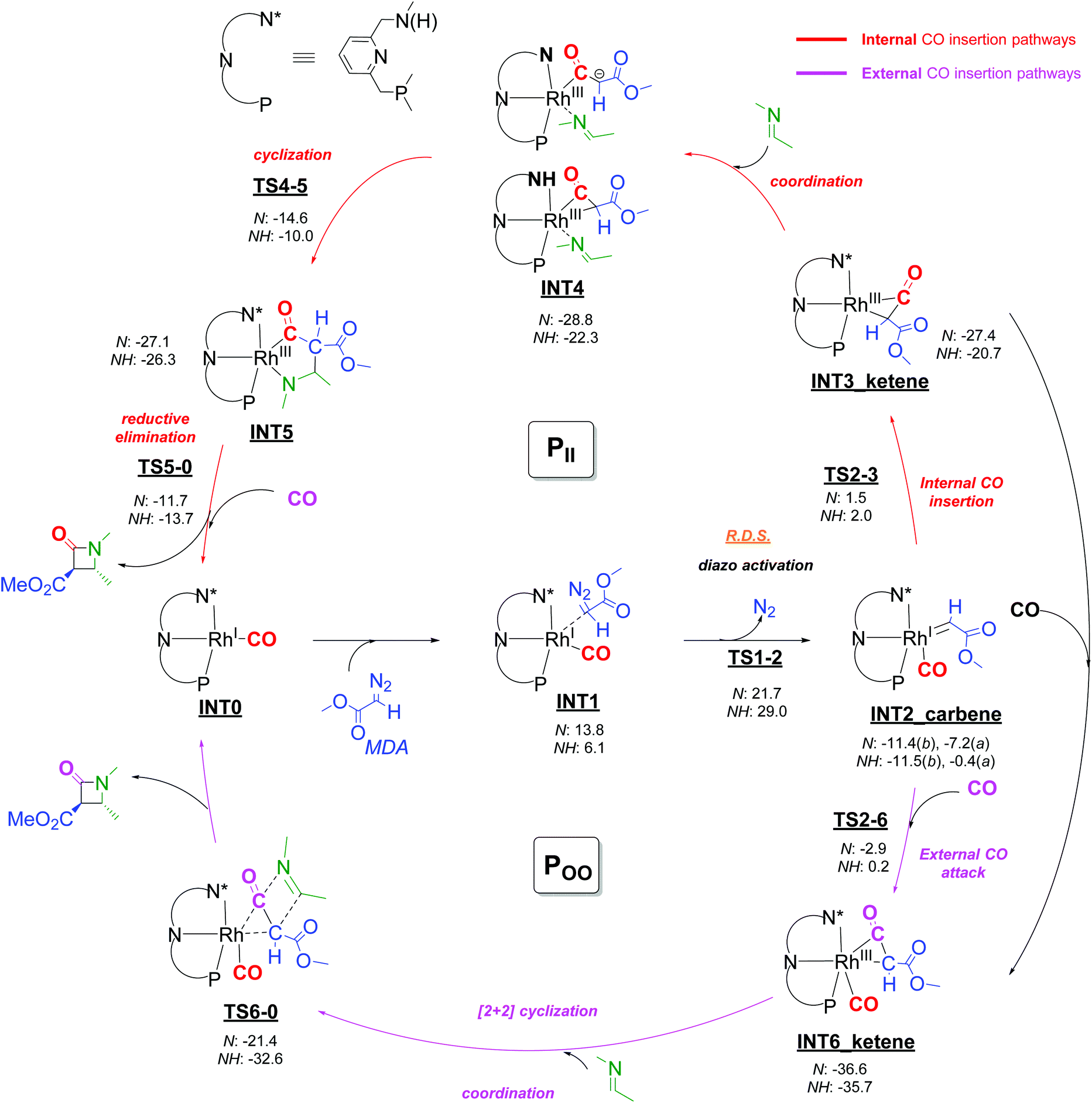 | ||
Scheme 7 DFT calculated (DFT-D3, BP86, def2-TZVP) pathways for β-lactam formation via catalytic generation of ketenes from rhodium-carbenoids (isodesmic, balanced reactions). Free energies (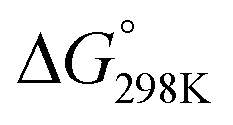 ) in kcal mol−1. All energies (also those of the transition states) are reported relative to species INT0. *N and NH denote the amido and amino forms, respectively, of the intermediates. The metal complexes are correspondingly neutral and cationic, the labels were omitted for clarity. *For INT2, ‘b’ indicates the carbene moiety is in a basal position; ‘a’ indicates the carbene moiety is in the axial position. ) in kcal mol−1. All energies (also those of the transition states) are reported relative to species INT0. *N and NH denote the amido and amino forms, respectively, of the intermediates. The metal complexes are correspondingly neutral and cationic, the labels were omitted for clarity. *For INT2, ‘b’ indicates the carbene moiety is in a basal position; ‘a’ indicates the carbene moiety is in the axial position. | ||
The catalytically active Rh-PNN species may be present either in their neutral NH-deprotonated amido form, or in their cationic non-deprotonated amine form. Both possibilities were investigated. The pathways and species involving cationic Rh-complexes with neutral PNNH amine ligands are denoted with an NH label (NHP pathway), while the pathways and species involving neutral amido complexes with anionic (N-deprotonated) PNN− ligands are labelled with an N label (NP pathway), see Scheme 7. A truncated ligand model of the Rh(CO)(PNN) complexes was used in all cases to limit computational costs, with all the substituents at nitrogen and phosphorus being replaced by methyl groups. Methyl diazo acetate (MDA) was used as the model diazo-compound.
Metallocarbene generation from decomposition of the diazo species starts with coordination of MDA to the square planar Rh(CO)(PNN) complex at the apical position (INT0 → INT1). This step is endergonic for both the cationic species and the neutral species, but formation of the MDA adduct of the cationic amine complex is less endergonic (by −7.7 kcal mol−1) than for the neutral amido complex. In contrast, and somewhat unexpectedly, subsequent N2 elimination from the MDA adducts to give the five-coordinated metallocarbene species INT2 has a lower barrier (ΔGǂ = +21.7 kcal mol−1) for the neutral amido complex as compared to the cationic amine complex (ΔGǂ = +29.0 kcal mol−1), despite the weaker Rh-MDA interaction in the precursor complex (transition states TS1-2; see Scheme 7). The lower activation barrier for rhodium-carbenoid formation at the neutral amido complex is presumably due to the more electron-rich nature of the metal centre of the neutral complex relative to the cationic amine complex, which stabilizes the carbene complex due to enhanced metal-to-carbene π-back donation. Overall, the computed diazo-activation steps to form the carbene intermediates INT2 are similar to those proposed by Pirrung, Hengge, Wu and coworkers on the basis of experimental kinetic studies,32 and several computational studies on other systems have revealed similar transition states for transition metal carbenoid formation from diazo compounds.33
Several subsequent steps were considered in the DFT calculations that provide pathways to β-lactam formation, of which several have lower total barriers than TS1-2 (see Scheme 7 and descriptions below), implying that diazo activation must be the overall rate limiting step of the catalytic cycle. The lower barrier for rhodium-carbenoid formation at the neutral amido species is in agreement with the experimental studies, showing that the use of basic conditions (either by using K2CO3 or using basic tosyl hydrazone salts as substrates) is beneficial.
Two isomeric forms of the carbene species INT2 differing in the position of the carbene ligand (in basal (b) and apical (a) positions) were considered (Fig. 2). For both the neutral amido and the cationic amine species, the carbene species INT2b having the carbene in the basal position are significantlly more stable than the carbene species INT2a having the carbene moiety in the apical position. Subsequent steps are considered to proceed from the lowest energy intermediate INT2b.
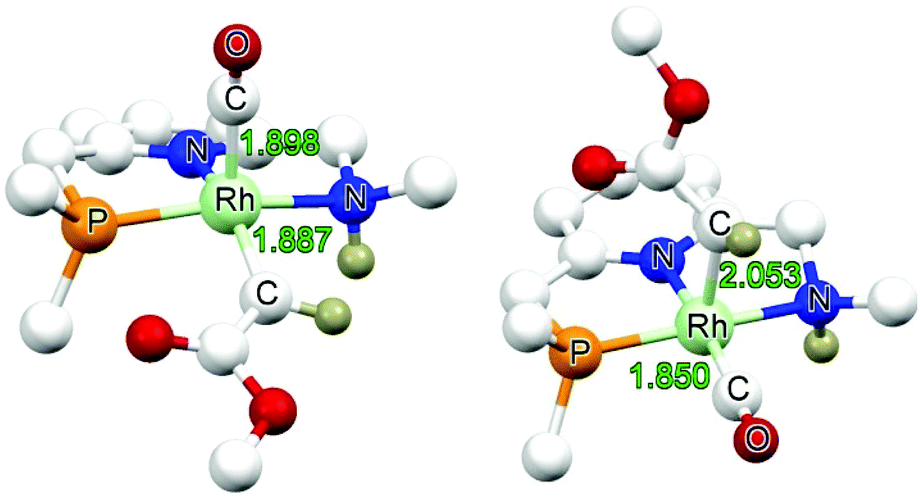 | ||
| Fig. 2 DFT optimized structure of, left: NHINT2b, right: NHINT2a (bond lengths in Å). | ||
Since the applied empirical dispersion corrections might be somewhat overestimated34 and at the same time the computed entropy effect of the two processes are widely different with one process being intramolecular and the other being intermolecular (for which computed entropy factors are overestimated), any reliable distinction between these inner-sphere and outer-sphere pathways can probably not be made on the basis of DFT. Therefore both inner- and outer-sphere pathways for formation of the (coordinated) ketene can be considered viable, and hence both computed pathways are discussed below.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C moiety is “softer” than the CO unit.36 The amido ketene species is 6.7 kcal mol−1 more stable than the amine derivative, in agreement with enhanced metal-to-ligand back-donation in the former.
C moiety is “softer” than the CO unit.36 The amido ketene species is 6.7 kcal mol−1 more stable than the amine derivative, in agreement with enhanced metal-to-ligand back-donation in the former.
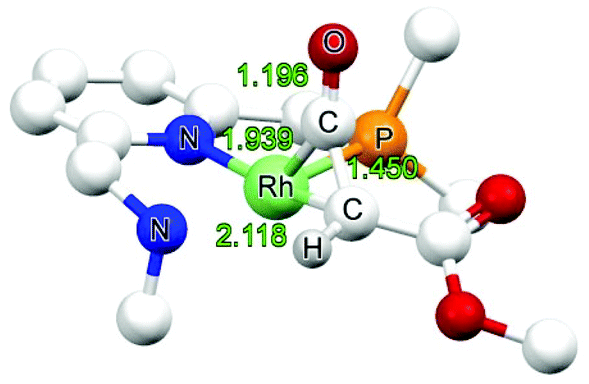 | ||
| Fig. 3 DFT optimized structure of NINT3 (bond lengths in Å). | ||
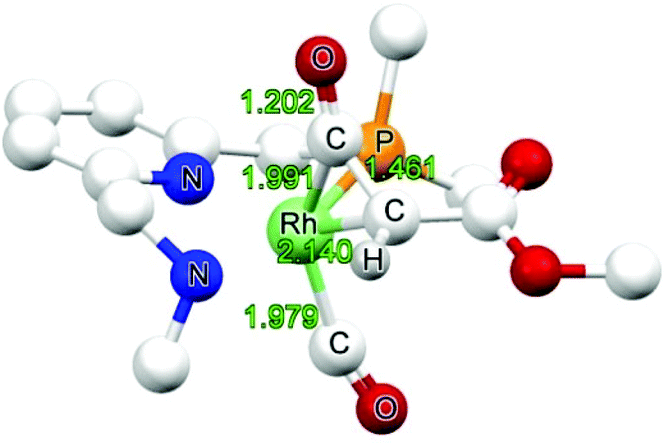 | ||
| Fig. 4 DFT optimized structure of NINT6 (bond lengths in Å). | ||
Two other isomeric forms were calculated to be slightly higher in energy, one being the Rh-η2-COketene isomer (+0.4 kcal mol−1 for the amido form) and the other having the CO coordinated trans to the pyridine and the ketene loosely bound at the axial position (+3.4 kcal for the amido form), suggesting possible ketene slippage and partial dissociation from the metal.
External attack of CO to INT2 to form ketene adduct INT6 is a process with a somewhat lower barrier (+8.5 and +11.7 kcal mol−1 for the amido and the amine forms, respectively) than the inner-sphere pathway to form INT3. Regarding the overestimation of the entropy penalty, these barriers may in reality be even lower. On the other hand the empirical dispersion effects in the approach of the CO ligand may be overestimated, and considering the rather small relative energy differences concerning these barriers, this makes it rather difficult to judge if the route viaTS2-3 or viaTS2-6 is the preferred pathway. Hence, we consider both pathways viable.
Follow-up reactivity of the ketene intermediates INT3 and INT6 with the imine substrate
One possible pathway leading to β-lactam formation after formation of the ketene adducts INT3 and INT6 is ketene dissociation followed by an uncatalysed coupling step between the free, uncoordinated ketene and the imine substrate. However, according to DFT the uncatalysed coupling of the free (uncoordinated) ketene and imine substrates (see Scheme 8) is associated with a very high barrier of 39.7 kcal mol−1.37,38 Importantly, the computed Rh-catalysed imine-ketene coupling steps in pathways PII and POO discussed below have much lower barriers (all computations performed using the same level of theory: DFT-D3, BP86, def2-TZVP), strongly suggesting that the final imine-ketene coupling steps of the β-lactam formation reaction are also Rh-catalysed.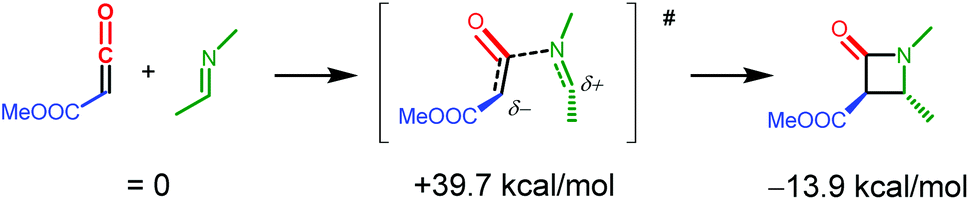 | ||
| Scheme 8 DFT computed (DFT-D3, BP86, def2-TZVP) uncatalysed coupling of the free (uncoordinated) ketene and imine substrates. | ||
From the neutral and cationic ketene intermediates INT3 and INT6 the possible pathways for Rh-catalysed β-lactam formation further diverge, and four different routes (partly interconnected by possible CO coordination/dissociation steps) were considered relevant for the formal imine [2 + 2] cyclization with the metal-bound ketene: The PII and PIO pathways each proceed from ketene intermediate INT3 and concern inner-sphere and outer-sphere pathways for coupling of the imine reagent with the coordinated ketene ligand, respectively. In the pathways PII the imine first coordinates to the metal before it couples to the ketene CC bond, whereas in the PIO pathway the imine directly attacks the coordinated ketene ligand as an external nucleophile. The POO and POI pathways each proceed from ketene intermediate INT6, and describe similar inner-sphere and outer-sphere pathways for coupling of the imine with the ketene ligand of INT6. These latter pathways are thus influenced by the presence of an additional CO ligand bound to rhodium. A short description of the two more plausible pathways PII and POO follows below, while the alternative, but energetically less favourable pathways PIO and POI are shown and discussed in some detail in the ESI (Scheme S1†).
Pathway PII: Coordination of the imine substrate to INT3 is slightly exergonic for both the cationic amine complex and the neutral amido species, producing intermediates INT4. For the neutral amido form of the imine-coordinated intermediate INT4 the ketene coordinates as an acetyl ligand with a κ1-Cα coordination pattern, while in the cationic amine form of INT4 the Rh-η2-CCketene-coordination mode is maintained. Intramolecular ring-closure viaTS4-5 involving C–C bond formation converts INT4 into the five-membered metallocyclic intermediates INT5, which produce the β-lactam product after successive reductive cyclization (TS5-0). For both the cationic amine and the neutral amido species, the TS4-5 (N: +14.2 kcal mol−1; NH: +12.3 kcal mol−1) and TS5-0 (N: +15.4 kcal mol−1; NH: +12.6 kcal mol−1) free energy barriers are very accessible, but the barriers for the cationic amine species are slightly lower than those of the neutral amido derivatives. In both cases the barriers for this pathway are much lower than the total barrier of the rate limiting diazo activation step (N: +21.7 kcal mol−1; NH: +29.0 kcal mol−1). Release of the product and association of CO regenerates the starting complex INT0, thus closing the catalytic cycle.
Pathway POO: The outer-sphere ketene-imine cyclization was calculated to occur in a concerted [2 + 2] step, directly at the coordinated ketene moiety of INT6 contrasting all other pathways involving stepwise couplings. This concerted reaction step (INT6 → TS6-0 → INT0 + β-lactam) directly produces the β-lactam product with regeneration of the starting complex INT0. It has a very low barrier for the cationic amine species (ΔGǂ = +3.1 kcal mol−1), while it has a significant but nonetheless very accessible barrier for the neutral amido species (ΔGǂ = 15.2 kcal mol−1). Considering that the rate limiting diazo activation step viaTS1-2 has a lower barrier for the deprotonated form of the catalyst, the barrier for the neutral amido species seems however most relevant in this POO pathway. This reaction pathway is a viable route for catalytic β-lactam product formation, especially considering the relative stability of intermediate INT6 as compared to INT3.
Pathway PIO and POI: Two alternative pathways PIO and POI were explored (see Scheme S1 in the ESI†), but both proceed via much higher barriers than pathways PII and POO. Pathway PIO proceeds via direct attack of the imine nitrogen atom at the ketene moiety of INT3 followed by stepwise ring closing process, with exceedingly high overall barriers from INT3 (N: INT3 → TS8-0, ΔGǂ = +40.8 kcal mol−1; NH: INT6 → TS8-0, ΔGǂ = +32.6 kcal mol−1), making this route much less favourable than pathway PII. For pathway POI, the first transition state for stepwise ring closing process represents rather high overall barriers from intermediate INT6 (INT6 → TS9-10: N: ΔGǂ = +25.3 kcal mol−1; NH: ΔGǂ = +19.6 kcal mol−1), which is clearly less favorable than pathway POO. For more details about pathway PIO and POI, see the ESI.†
Brief summary of the DFT calculations
The combined DFT calculations can be summarized as follows: The activation of the diazo compound to form the carbene intermediate INT2 appears to be the rate-limiting step of the reaction, and this step is faster upon amine deprotonation of the PNN ligand of the catalyst. Hence (in agreement with the experimental observations), it seems that the reactions preferably take place with the catalyst in its amine deprotonated form. As such, in the remaining discussion below the energetic barriers listed are limited to those concerning the most relevant neutral amido species.From INT2, the saturated and unsaturated ketene intermediates INT6 and INT3 can be formed, with INT6 calculated to be lower in energy. From INT3 pathway PII is the lowest barrier route to β-lactam formation, while for INT6 the lowest barrier route is the POO pathway (ΔGǂ = +15.2 kcal mol−1). Considering the higher stability of INT6, the overall barriers along the PII pathway are somewhat higher than along the POO pathway (ΔGǂ = +22.0 kcal mol−1versus +15.2 kcal mol−1). However, overestimated dispersion corrections could play a role in favoring the POO pathway over the PII pathway, and hence both pathways remain viable routes to β-lactam formation. The PIO and POI pathways can be safely excluded based on their relatively overall energy barriers as compared to the PII and POO pathways, respectively (see Scheme S1†).
Stoichiometric experimental studies aimed at showing formation of ketenes by internal CO attack
In order to get experimental information for ketene formation, we first investigated the possibility for internal CO migration to the Rh-carbenoid moiety to form the ketene product. When two equivalents of EDA (1) were added to a CD2Cl2 solution of complex B at room temperature, no reaction except for dimerization of the diazo-evolved carbenoid was observed in an overnight experiment (two isomers of diethyl fumarate giving two sharp singlets at 6.2 and 6.8 ppm, respectively, see ESI, Fig. S2†). When two equivalents of N-tosyl hydrazone salt 5 were added to complex B in C6D6, complex B was immediately deprotonated, accompanied with a colour change from yellow to purple-red. Heating the solution to 55 °C for 1 hour did not produce any phenylketene. In another reaction, one equivalent of DBU was added to deprotonate the NH moiety of the PNN ligand of complex B prior to the addition of the N-tosyl hydrazone salt 5, to avoid generation of the organic N-tosyl hydrazone precursor, which would interfere in the 1H NMR spectrum. Heating the reaction mixture to 55 °C for five hours gave rise to a singlet at 4.88 ppm in the 1H NMR spectrum (see ESI, Fig. S4 and S5†), which indeed indicates formation of the ketene species. Other signals are slightly broadened and hence not clear. In the 31P NMR spectrum, one doublet at 49.85 ppm with a small JRhP coupling constant of 137.8 Hz suggested that the PNN ligand is probably in the amido form.39 This result indicates that inner-sphere CO insertion is indeed a viable possibility under the applied reaction conditions to form the ketene fragment, but whether or not this process is truly involved in the catalytic routes is less clear.Conclusions
In summary, we have synthesized a series of rhodium complexes featuring new secondary amine containing PNN ligands. Complex B and E showed good activity in catalytic ketene synthesis involving carbonylation of carbenes. Amides and β-lactams can be synthesized in one-pot reactions proceeding via in situ generated ketene intermediates. A ketene imine has also been synthesized in a catalytic manner by reaction of a carbenoid with an isocyanide.DFT computational studies suggest that activation of the diazo compound to generate the rhodium-carbenoid intermediate is the rate-determining step of the catalytic reaction, with the neutral amido complex being more reactive than the cationic amine complex. This is in agreement with the experimental studies, showing that the use of basic conditions (either by using K2CO3 or using basic tosyl hydrazone salts as substrates) is beneficial. Outer-sphere CO insertion is computed to be slightly favoured over the inner-sphere mechanism. Preliminary experimental studies show that inner sphere CO insertion to form ketene is also possible, but whether or not this process is truly involved in the catalytic routes is less clear. Additional spectrometric and kinetic studies are needed to unveil further details about the mechanism of these reactions. Follow-up coupling mechanisms most likely proceed via either the POO or PII pathways depicted in Scheme 7, with the POO pathway being slightly favoured over the PII pathway according to DFT.
Experimental details
General methods
All reactions were carried out under an argon atmosphere using standard Schlenk techniques or in a glovebox unless noted otherwise. With exception of the compounds given below, all reagents were purchased from commercial suppliers and used without further purification. N-Methyl-1-(6-methylpyridin-2-yl)methanamine (for synthesis of a)40 and N-((6-methylpyridin-2-yl)methyl)aniline (for synthesis of b)41 were synthesized from 6-methylpicolinaldehyde according to literature procedures. 2-(Chloromethyl)-6-((di-tert-butylphosphino)-methyl)pyridine·BH3 (for the synthesis of d) and 2-(chloromethyl)-6-((diisopropylphosphino)methyl)pyridine·BH3 (for the synthesis of c and e) were synthesized from 2,6-bis(chloromethyl)pyridine according to a reported procedure.42 Complex D was synthesized according to a reported literature procedure.22 THF, dioxane, pentane, and diethyl ether were distilled from sodium (benzophenone as indicator) under nitrogen. CH2Cl2 and methanol were distilled from CaH2 under nitrogen. N-Tosylhydrazone sodium salts were synthesized according to published procedures.43,44 6-Methylpicolinaldehyde and 2,6-bis(chloromethyl)pyridine were purchased from TCI Europe. Picolinaldehyde and ethyl diazoacetate (EDA) were purchased from Aldrich (up to 15% dichloromethane, actual content determined by NMR). NMR spectra (1H, 31P{1H} and 13C{1H}) were measured on a Bruker AMX 400 spectrometer at 25 °C, unless noted otherwise. IR spectra were recorded on a Nicolet Nexus FT-IR spectrometer. HR-MS were obtained using a time-of-flight JEOL AccuTOF LC-plus mass spectrometer (JMS-T100LP).
In situ ketene synthesis by carbene carbonylation
In a typical carbonylation experiment, a stainless steel autoclave (150 mL) suitable for seven reaction vessels (equipped with Teflon mini-stirring bars) was used to perform parallel reactions. Each vial was charged with carbene precursor (EDA 1 or N-tosylhydrazone sodium salt 5, 0.15 mmol), amine nucleophile (0.30 mmol), rhodium catalyst (0.0075 mmol), internal standard 1,3,5-trimethoxybenzene (0.015 mmol) and solvent (3 mL). Before starting the catalytic reactions, the charged autoclave was purged (5×) with 20 bar of CO and then pressurized to the desired pressure. The autoclave was heated in an oil-bath with the vials being stirred overnight. After the reaction, the autoclave was cooled to 0 °C and vented to 1 atm. Around 1.5 mL was taken out of each reaction mixture, and the solvent was removed via rotary evaporation. The residue was dissolved in CDCl3 or CH2Cl2 (and filtered when necessary) for 1H NMR and GC-MS analysis, respectively. The conversion and yield for each reaction were calculated based on integration of the 1H NMR signals of the corresponding species relative to that of the internal standard. Product assignment followed assignments presented in literature.14eKetene imine synthesis involving carbene–isocyanide coupling
A 25 mL Schlenk vessel was charged with N-tosylhydrazone sodium salt 5 (0.15 mmol), rhodium catalyst (0.0075 mmol), internal standard 1,3,5-trimethoxybenzene (0.015 mmol), toluene (5 mL) and tert-butyl isocyanide (0.75 mmol). The Schlenk vessel was then sealed and heated in an oil-bath overnight. After the reaction, around 2.5 mL was taken out of the reaction mixture, and the solvent was removed via rotary evaporation. The residue was dissolved in CDCl3 or CH2Cl2 and filtered for 1H NMR and GC-MS analysis, respectively. The conversion and yield for each reaction were calculated based on integration of the 1H NMR signals of the corresponding species relative to that of the internal standard.X-ray crystal structure determination of complex A
Reflections were measured on a Bruker Kappa ApexII diffractometer with sealed tube and Triumph monochromator (λ = 0.71073 Å) at a temperature of 150(2) K up to a resolution of (sin θ/λ)max = 0.81 Å−1. The intensities were integrated with the Eval15 software.45 Multiscan absorption correction and scaling was performed with SADABS46 (correction range 0.67–0.75). The structure was solved with Patterson superposition methods using SHELXT.47 Least-squares refinement was performed with SHELXL-97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 48 against F2 of all reflections. Non-hydrogen atoms were refined freely with anisotropic displacement parameters. The coordinated amino nitrogen was disordered over two positions with a ratio 0.884(6):0.116(6). All hydrogen atoms were located in difference Fourier maps. The hydrogen atom at N2 was refined freely with an isotropic displacement parameter, all other H-atoms were refined with a riding model. Geometry calculations and checking for higher symmetry was performed with the PLATON program.49
48 against F2 of all reflections. Non-hydrogen atoms were refined freely with anisotropic displacement parameters. The coordinated amino nitrogen was disordered over two positions with a ratio 0.884(6):0.116(6). All hydrogen atoms were located in difference Fourier maps. The hydrogen atom at N2 was refined freely with an isotropic displacement parameter, all other H-atoms were refined with a riding model. Geometry calculations and checking for higher symmetry was performed with the PLATON program.49
X-ray crystal structure details of complex A
[C21H21N2OPRh](PF6)·CH3OH, Fw = 628.29, yellow block, 0.57 × 0.35 × 0.18 mm3, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 9.70447(19), b = 9.77001(15), c = 14.1603(3) Å, α = 83.358(1), β = 74.636(1), γ = 74.636(1)°, V = 1246.84(4) Å3, Z = 2, Dx = 1.674 g cm−3, μ = 0.88 mm−1. 41801 reflections were measured up to a resolution of (sinθ/λ)max = 0.81 Å−1. 10952 reflections were unique (Rint = 0.015), of which 10239 were observed [I > 2σ(I)]. 330 parameters were refined with 1 restraint (C–N distance of the disordered group). R1/wR2 [I > 2σ(I)]: 0.0221/0.0567. R1/wR2 [all refl.]: 0.0243/0.0577. S = 1.057. Residual electron density between −0.53 and 0.61 e Å−3. CCDC 1424620.
(no. 2), a = 9.70447(19), b = 9.77001(15), c = 14.1603(3) Å, α = 83.358(1), β = 74.636(1), γ = 74.636(1)°, V = 1246.84(4) Å3, Z = 2, Dx = 1.674 g cm−3, μ = 0.88 mm−1. 41801 reflections were measured up to a resolution of (sinθ/λ)max = 0.81 Å−1. 10952 reflections were unique (Rint = 0.015), of which 10239 were observed [I > 2σ(I)]. 330 parameters were refined with 1 restraint (C–N distance of the disordered group). R1/wR2 [I > 2σ(I)]: 0.0221/0.0567. R1/wR2 [all refl.]: 0.0243/0.0577. S = 1.057. Residual electron density between −0.53 and 0.61 e Å−3. CCDC 1424620.
X-ray crystal structure determination of complexes B and E
X-ray intensities were measured on a Bruker D8 Quest Eco diffractometer equipped with a Triumph monochromator (λ = 0.71073 Å) and a CMOS Photon 50 detector at a temperature of 150(2) K. Intensity data were integrated with the Bruker APEX2 software.50 Absorption correction and scaling was performed with SADABS.46 The structures were solved with the program SHELXL.50 Least-squares refinement was performed with SHELXL-201351 against F2 of all reflections. Non-hydrogen atoms were refined with anisotropic displacement parameters. The H atoms were placed at calculated positions using the instructions AFIX 13, AFIX 43 or AFIX 137 with isotropic displacement parameters having values 1.2 or 1.5 times Ueq of the attached C atoms.
X-ray crystal structure details of complex B
C26H23N2OPRh PF6, Fw = 658.31, yellow block, 0.389 × 0.319 × 0.112 mm3, Monoclinic, P21/c, a = 10.6439(13), b = 11.6245(14), c = 21.782(2) Å, β = 100.416(2)°, V = 2650.7(5) Å3, Z = 4, Dx = 1.650 g cm−3, μ = 0. 831 mm−1. 30389 reflections were measured up to a resolution of (sinθ/λ)max = 0.59 Å−1. 4661 Reflections were unique (Rint = 0.0711), of which 3905 were observed [I > 2σ(I)]. 346 Parameters were refined with 334 restraints (RIGU). R1/wR2 [I > 2σ(I)]: 0.0382/0.0948. R1/wR2 [all refl.]: 0.0510/0.1062. S = 1.038. CCDC 1423805.
X-ray crystal structure details of complex E
C20H27N2OPRh PF6, Fw = 590.29, yellow-brown block, 0.886 × 0.217 × 0.126 mm3, monoclinic, P21/n, a = 13.4021(9), b = 10.9937(7), c = 15.6906(10) Å, β = 97.462(3)°, V = 2292.3(3) Å3, Z = 4, Dx = 1.710 g cm−3, μ = 0. 949 mm−1. 97401 reflections were measured up to a resolution of (sinθ/λ)max = 0.91 Å−1. 14600 reflections were unique (Rint = 0.0669), of which 10763 were observed [I > 2σ(I)]. 297 Parameters were refined with 279 restraints (RIGU). R1/wR2 [I > 2σ(I)]: 0.0381/0.0833. R1/wR2 [all refl.]: 0.0675/0.0988. S = 1.075. CCDC 1423806.
CCDC 1424620, 1423805 and 1423806 contain the supplementary crystallographic data for compound A, B and E, respectively.
Details of the DFT calculations
Geometry optimizations were carried out with the Turbomole program package52 coupled to the PQS Baker optimizer53via the BOpt package,54 at the ri-DFT level using the BP8655 functional and the resolution-of-identity (ri) method.56 The geometries of all stationary points were optimized at the def2-TZVP basis set level,57 both with and without Grimme's dispersion corrections (disp3 version).58 The identity of the transition states was confirmed by following the imaginary frequency in both directions (IRC). All minima (no imaginary frequencies) and transition states (one imaginary frequency) were characterized by calculating the Hessian matrix. ZPE and gas-phase thermal corrections (entropy and enthalpy, 298 K, 1 bar) from these analyses were calculated. The relative (free) energies obtained from these calculations are reported in Scheme 7.Acknowledgements
This research was funded by the Netherlands Organization for Scientific Research, section Chemical Sciences (NWO-CW; ECHO grant to BdB). JIvdV thanks the ERC for a Starting Grant (279097, EuReCat). The ApexII diffractometer at Utrecht University has been financed by NWO-CW. We thank Ed Zuidinga for performing the ESI-MS measurements.Notes and references
- (a) H. Staudinger, Ber. Dtsch. Chem. Ges., 1905, 38, 1735 CrossRef PubMed; (b) T. T. Tidwell, Eur. J. Org. Chem., 2006, 563 CrossRef CAS PubMed; (c) D. H. Paull, A. Weatherwax and T. Lectka, Tetrahedron, 2009, 65, 6771 CrossRef CAS PubMed.
- (a) A. D. Allen and T. T. Tidwell, Eur. J. Org. Chem., 2012, 1081 CrossRef CAS PubMed; (b) A. D. Allen and T. T. Tidwell, Chem. Rev., 2013, 113, 7287 CrossRef CAS PubMed.
- (a) H. Staudinger and J. Meyer, Chem. Ber., 1920, 53, 72 CrossRef PubMed; (b) H. Perst, in Science of Synthesis, ed. R. L. Danheiser, Product Class 17: Ketenimines, 781, Georg Thieme Verlag, Stuttgart, 2006, vol. 23 Search PubMed; (c) S. H. Kim, S. H. Park, J. H. Choi and S. Chang, Chem. – Asian J., 2011, 6, 2618 CrossRef CAS PubMed; (d) S. E. Denmark and T. W. Wilson, Angew. Chem., Int. Ed., 2012, 51, 9980 CrossRef CAS PubMed; (e) P. Lu and Y. Wang, Chem. Soc. Rev., 2012, 41, 5687 RSC; (f) C. Fromont and S. Masson, Tetrahedron, 1999, 55, 5405 CrossRef CAS; (g) G. A. Gornowicz and R. West, J. Am. Chem. Soc., 1971, 93, 1714 CrossRef; (h) I. Bae, H. Han and S. Chang, J. Am. Chem. Soc., 2005, 127, 2038 CrossRef CAS PubMed.
- H. W. Moore and D. S. Wilbur, J. Org. Chem., 1980, 45, 4483 CrossRef CAS.
- (a) Y. Chiang, A. J. Kresge and V. V. Popik, J. Am. Chem. Soc., 1999, 121, 5930 CrossRef CAS; (b) B. D. Wagner, B. R. Arnold, G. S. Brown and J. Lusztyk, J. Am. Chem. Soc., 1998, 120, 1827 CrossRef CAS.
- L. S. Hegedus, J. Montgomery, Y. Narukawa and D. C. Snustad, J. Am. Chem. Soc., 1991, 113, 5784 CrossRef CAS.
- Y. Yang, W. Shou, D. Hong and Y. Wang, J. Org. Chem., 2008, 73, 3574 CrossRef CAS PubMed.
- (a) G. A. Gornowicz and R. West, J. Am. Chem. Soc., 1971, 93, 1714 CrossRef; (b) M. Bendikov, H. M. Duong, E. Bolanos and F. Wudl, Org. Lett., 2005, 7, 783 CrossRef CAS PubMed; (c) Y. Zhang, K. A. DeKorver, A. G. Lohse, Y. Zhang, J. Huang and R. P. Hsung, Org. Lett., 2009, 11, 899 CrossRef CAS PubMed; (d) K. A. DeKorver, X. N. Wang, M. C. Walton and R. P. Hsung, Org. Lett., 2012, 14, 1768 CrossRef CAS PubMed.
- (a) M. P. Cassidy, J. Raushel and V. V. Fokin, Angew. Chem., Int. Ed., 2006, 45, 3154 CrossRef CAS PubMed; (b) M. Whiting and V. V. Fokin, Angew. Chem., Int. Ed., 2006, 45, 3157 CrossRef CAS PubMed.
- (a) N. Ungvári and F. Ungváry, Carbonylation of diazoalkanes, in Modern Carbonylation Methods, ed. L. Kollár, Wiley-VCH, Weinheim, 2008, ch. 8, p. 199 Search PubMed; (b) K. H. Dötz and J. Stendel, Chem. Rev., 2009, 109, 3227 CrossRef PubMed.
- H. Staudinger and O. Kupfer, Ber. Dtsch. Chem. Ges., 1912, 45, 501 CrossRef CAS PubMed.
- (a) K. H. Dötz and J. Mühlemeier, Angew. Chem., Int. Ed. Engl., 1982, 21, 929 CrossRef PubMed; (b) P. T. Barger, B. D. Santarsiero, J. Armantrout and J. E. Bercaw, J. Am. Chem. Soc., 1984, 106, 5178–5186 CrossRef CAS; (c) N. Ungvári, E. Fördős, J. Balogh, T. Kégl, L. Párkányi and F. Ungváry, Organometallics, 2010, 29, 3837 CrossRef.
- TM-catalysed carbonylation of dihalides: (a) A. Miyashita, T. Kihara, K. Nomura and H. Nohira, Chem. Lett., 1986, 1607 CrossRef CAS; (b) M. Huser, M. Youinou and J. A. Osborn, Angew. Chem., Int. Ed. Engl., 1989, 28, 1386 CrossRef PubMed.
- TM-catalysed carbonylation of diazo compounds: (a) T. Ye and M. A. Mckervey, Chem. Rev., 1994, 94, 1091 CrossRef CAS; (b) R. Tuba and F. Ungváry, J. Mol. Catal. A: Chem., 2003, 203, 59 CrossRef CAS; (c) Z. Zhang and J. Wang, Tetrahedron, 2008, 64, 6577 CrossRef CAS PubMed; (d) E. Fördos, R. Tuba, L. Párkányl, T. Kégl and F. Ungváry, Eur. J. Org. Chem., 2009, 1994 CrossRef PubMed; (e) Z. Zhang, Y. Liu, L. Ling, Y. Li, Y. Dong, M. Gong, X. Zhao, Y. Zhang and J. Wang, J. Am. Chem. Soc., 2011, 133, 4330 CrossRef CAS PubMed; (f) N. D. Paul, A. Chirila, H. Lu, X. P. Zhang and B. de Bruin, Chem. – Eur. J., 2013, 19, 12953 CrossRef CAS PubMed; (g) Z. Zhang, Y. Zhang and J. Wang, ACS Catal., 2011, 1, 1621 CrossRef CAS; (h) Q. Xiao, Y. Zhang and J. Wang, Acc. Chem. Res., 2013, 46, 236 CrossRef CAS PubMed.
- J. R. Fulton, V. K. Aggarwal and J. De Vicente, Eur. J. Org. Chem., 2005, 1479 CrossRef CAS PubMed.
- C. R. Pitts and T. Lectka, Chem. Rev., 2014, 114, 7930 CrossRef CAS PubMed.
- (a) J. A. Green and L. A. Singer, Tetrahedron Lett., 1969, 5093 CrossRef CAS; (b) T. W. Hudnall, E. J. Moorhead, D. G. Gusev and C. W. Bielawski, J. Org. Chem., 2010, 75, 2763 CrossRef CAS PubMed; (c) G. A. Blake, J. P. Moerdyk and C. W. Bielawski, Organometallics, 2012, 31, 3373 CrossRef CAS; (d) D. Martin, Y. Canac, V. Lavallo and G. Bertrand, J. Am. Chem. Soc., 2014, 136, 5023 CrossRef CAS PubMed.
- (a) F. Zhou, K. Ding and Q. Cai, Chem. – Eur. J., 2011, 17, 12268 CrossRef CAS PubMed; (b) Q. Dai, Y. Jiang, J.-T. Yu and J. Cheng, Chem. Commun., 2015 10.1039/c5cc06771e.
- R. Lindner, B. van den Bosch, M. Lutz, J. N. H. Reek and J. I. van der Vlugt, Organometallics, 2011, 30, 499 CrossRef CAS.
- (a) E. Fogler, J. A. Garg, P. Hu, G. Leitus, L. J. W. Shimon and D. Milstein, Chem. – Eur. J., 2014, 20, 15727 CrossRef CAS PubMed.
- C. Tejel, M. P. Río, L. Asensio, F. J. Van Den Bruele, M. A. Ciriano, N. Tsichlis i Spithas, D. G. H. Hetterscheid and B. de Bruin, Inorg. Chem., 2011, 50, 7524 CrossRef CAS PubMed.
- For complex D, see: Z. Tang, E. Otten, J. N. H. Reek, J. I. van der Vlugt and B. de Bruin, Chem. – Eur. J., 2015, 21, 12683–12693 CrossRef CAS PubMed.
- N2 of complex A is distorted over two positions with a ratio of 0.88:0.12, see ESI.†.
- Addition of K2CO3 (22.2 mg, 0.161 mmol) to a solution of B (27.1 mg, 0.041 mmol) in THF (1.5 mL) led to immediate color change from yellow to orange-red. The 31P NMR spectrum of the reaction solution showed a doublet at 49.58 (1JRhP = 141.5). The high-field chemical shift and remarkable decrease in coupling constant are indicative of NH deprotonation to generate a strong donor trans to phosphorus.
- (a) J.-M. Lehn, Chem. – Eur. J., 1999, 5, 2455–2463 CrossRef CAS; (b) M. Ciaccia and S. Di Stefano, Org. Biomol. Chem., 2015, 13, 646–654 RSC.
- Alternatively, 7a could be formed by direct coupling of the metallocarbene intermediate II and the aniline nucleophile, followed by a parallel dehydrogenation process. However, no dehydrogenation or transfer hydrogenation of benzylamine was observed when using B′ or E′ as the catalyst under similar conditions, indicating a fast dehydrogenation process of a metal-bound amine/amido intermediate following the undesired C–N coupling, if that is the case.
- (a) R. B. Morin and M. Gorman, Chemistry and Biology of β-Lactam Antibiotics, Academic Press, New York, 6th edn, 1982 Search PubMed; (b) B. Alcaide, P. Almendros and C. Aragoncillo, Chem. Rev., 2007, 107, 4437 CrossRef CAS PubMed; (c) T. T. Tidwell, Angew. Chem., Int. Ed., 2008, 47, 1016 CrossRef CAS PubMed; (d) N. A. A. El-Kanzi, Heterocycl. Lett., 2013, 3, 81 Search PubMed; (e) A. Gupta and A. K. Halve, Int. J. Pharm. Sci. Res., 2015, 6, 978 CAS.
- (a) M. Staudinger, Justus Liebigs Ann. Chem., 1907, 356, 51 CrossRef PubMed; (b) G. I. Georg and V. T. Ravikumar, in The Organic Chemistry of β-Lactams, ed. G. I. Georg, Verlag Chemie, New York, 1993, p. 295 Search PubMed; (c) C. Palomo, J. M. Aizpurua, I. Ganboa and M. Oiarbide, Curr. Med. Chem., 2004, 11, 1837 CrossRef CAS; (d) R. Tuba, Org. Biomol. Chem., 2013, 11, 5976 RSC.
- (a) L. S. Hegedus, J. Montgomery, Y. Narukawa and D. C. Snustad, J. Am. Chem. Soc., 1991, 113, 5784 CrossRef CAS; (b) S. France, A. Weatherwax, A. E. Taggi and T. Lectka, Acc. Chem. Res., 2004, 37, 592 CrossRef CAS PubMed; (c) Y. Wang, Y. Liang, L. Jiao, D. M. Du and J. Xu, J. Org. Chem., 2006, 71, 6983 CrossRef CAS PubMed; (d) B. K. Banik, B. Lecea, A. Arrieta, A. De Cózar and F. P. Cossío, Angew. Chem., Int. Ed., 2007, 46, 3028 CrossRef CAS PubMed; (e) F. P. Cossío, A. Arrieta and M. A. Sierra, Acc. Chem. Res., 2008, 41, 925 CrossRef PubMed; (f) H. Qi, X. Li and J. Xu, Org. Biomol. Chem., 2011, 9, 2702 RSC.
- R. Tuba, Org. Biomol. Chem., 2013, 11, 5976 CAS.
- (a) A. Arrieta, F. P. Cossı, I. Ferna and M. J. Manchen, J. Am. Chem. Soc., 2000, 122, 11509 CrossRef CAS; (b) I. Ferna, M. a. Sierra, J. Manchen, M. Go and F. P. Cossi, J. Am. Chem. Soc., 2008, 130, 13892 CrossRef PubMed.
- (a) M. C. Pirrung, H. Liu and A. T. Morehead Jr., J. Am. Chem. Soc., 2002, 124, 1014 CrossRef CAS PubMed; (b) F. M. Wong, J. Wang, A. C. Hengge and W. Wu, Org. Lett., 2007, 9, 1663 CrossRef CAS PubMed.
- (a) E. Nakamura, N. Yoshikai and M. Yamanaka, J. Am. Chem. Soc., 2002, 124, 7181 CrossRef CAS PubMed; (b) J. Hansen, J. Autschbach and H. M. L. Davies, J. Org. Chem., 2009, 74, 6555 CrossRef CAS PubMed; (c) N. D. Paul, S. Mandal, M. Otte, X. Cui, X. P. Zhang and B. de Bruin, J. Am. Chem. Soc., 2014, 136, 1090 CrossRef CAS PubMed; (d) W. I. Dzik, X. Xu, X. P. Zhang, J. N. H. Reek and B. de Bruin, J. Am. Chem. Soc., 2010, 132, 10891 CrossRef CAS PubMed; (e) A. J. C. Walters, J. N. H. Reek and B. de Bruin, ACS Catal., 2014, 4, 1376 CrossRef CAS.
- (a) T. Schwabe and S. Grimme, Phys. Chem. Chem. Phys., 2007, 9, 3397 RSC; (b) K. E. Riley and M. Piton, Chem. Rev., 2010, 110, 5023 CrossRef CAS PubMed; (c) J. P. P. Ramalho, J. R. B. Gomes and F. Illas, RSC Adv., 2013, 3, 13085 RSC.
- (a) R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533 CrossRef CAS; (b) R. G. Pearson, Science, 1966, 151, 172 CAS; (c) R. G. Pearson, Chemical Hardness - Applications From Molecules to Solids, Wiley-VCH, Weinheim, 1997 Search PubMed.
- (a) H. Werner and E. Bleuel, Angew. Chem., Int. Ed., 2001, 40, 145 CrossRef CAS; (b) D. B. Grotjahn, L. S. B. Collins, M. Wolpert, G. a. Bikzhanova, H. C. Lo, D. Combs and J. L. Hubbard, J. Am. Chem. Soc., 2001, 123, 8260 CrossRef CAS PubMed.
- The direct uncatalysed concerted (but non-synchronous) [2 + 2] cycloaddition reaction of the trans-imine MeCNMe and ketene OCCHCOOMe in the gas phase was considered, without any proton- or Lewis-acid-assistance (i.e. not acid-catalysed). This barrier seems most relevant for comparison with the Rh-catalysed pathways considering the fact that the experimental reactions were performed under basic reaction conditions.
- Rather high reaction barriers were also obtained in other studies of [2 + 2] cyclo-addition reactions involving several differently substituted ketenes and imines and using various computational models: (a) B. K. Banik, B. Lecea, A. Arrieta, A. de Cózar and F. P. Cossío, Angew. Chem., Int. Ed., 2007, 46, 3028 CrossRef CAS PubMed; (b) N. Fu and T. T. Tidwell, Tetrahedron, 2008, 64, 10465 CrossRef CAS PubMed; (c) C. Zhou, Studies on cycloaddition reactions of ketenes: further investigations of pseudopericyclic reaction mechanisms, PhD Thesis, Texas Tech University, 2004, http://hdl.handle.net/2346/10697 Search PubMed.
- L. S. Jongbloed, B. de Bruin, J. N. H. Reek, M. Lutz and J. I. van der Vlugt, Chem. – Eur. J., 2015, 21, 7297 CrossRef CAS PubMed.
- M. Seitz, A. Kaiser, A. Tereshchenko, C. Geiger, Y. Uematsu and O. Reiser, Tetrahedron, 2006, 62, 9973 CrossRef CAS PubMed.
- G. J. P. Britovsek, J. England and A. J. P. White, Dalton Trans., 2006, 1399 RSC.
- M. Gargir, Y. Ben-David, G. Leitus, Y. Diskin-Posner, L. J. W. Shimon and D. Milstein, Organometallics, 2012, 31, 6207 CrossRef CAS.
- J. R. Fulton, V. K. Aggarwal and J. de Vicente, Eur. J. Org. Chem., 2005, 1479 CrossRef CAS PubMed.
- (a) P. Doppelt, J. Fischer and R. Webs, Inorg. Chem., 1984, 23, 2958–2962 CrossRef CAS; (b) R. L. Halterman and S. T. Jan, J. Org. Chem., 1991, 56, 5253 CrossRef CAS; (c) X. Xu, H. Lu, J. V. Ruppel, X. Cui, S. L. de Mesa, L. Wojtas and X. P. Zhang, J. Am. Chem. Soc., 2011, 133, 15292 CrossRef CAS PubMed.
- A. M. M. Schreurs, X. Xian and L. M. J. Kroon-Batenburg, J. Appl. Crystallogr., 2010, 43, 70 CrossRef CAS.
- G. M. Sheldrick, SADABS, Universität Göttingen, Germany, 2008 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148 CrossRef CAS PubMed.
- Bruker, APEX2 software, Madison WI, USA, 2014 Search PubMed.
- G. M. Sheldrick, SHELXL2013, University of Göttingen, Germany, 2013 Search PubMed.
- R. Ahlrichs, Turbomole Version 6.5, Theoretical Chemistry Group, University of Karlsruhe Search PubMed.
- PQS version 2.4, Parallel Quantum Solutions, Fayettevile, Arkansas, USA, 2001 CrossRef PubMed; the Baker optimizer is available separately from PQS upon request: I. Baker, J. Comput. Chem., 1986, 7, 385 CrossRef PubMed.
- P. H. M. Budzelaar, J. Comput. Chem., 2007, 28, 2226 CrossRef CAS PubMed.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS; (b) J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822 CrossRef.
- M. Sierka, A. Hogekamp and R. Ahlrichs, J. Chem. Phys., 2003, 118, 9136 CrossRef CAS PubMed.
- A. Schaefer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1424620, 1423805 and 1423806. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00287g |
| This journal is © the Partner Organisations 2015 |