
Organopolymerization of naturally occurring Tulipalin B: a hydroxyl-functionalized methylene butyrolactone†
Jing
Tang
and
Eugene Y.-X.
Chen
*
Department of Chemistry, Colorado State University, Fort Collins, Colorado 80523-1872, USA. E-mail: eugene.chen@colostate.edu
First published on 6th November 2015
Abstract
The first successful polymerization of the naturally occurring, OH-containing, tri-functional monomer Tulipalin B (βHMBL) is established. N-Heterocyclic carbene and phosphazene superbase catalysts effectively polymerize βHMBL into polymers with Mn up to 13.2 kg mol−1. The possible polymer structure is thought to be a branched copolymer of poly(vinyl-ether lactone)s, derived from proposed crossovers between conjugate Michael and oxa-Michael additions, enabled by proton transfer.
Introduction
The emergence of organopolymerization1 has profited from the development of powerful organic catalysts and unique mechanistic pathways in the rapidly growing field of organocatalysis.2 One such important class of organic catalysts is N-heterocyclic carbenes (NHCs); thanks to their inherently high Brønsted-basicity and nucleophilicity, NHCs have attracted increasing interest for their unique reactivity and selectivity often observed in many different types of organic reactions.3 The scope of the NHC-mediated polymerization4 has expanded significantly from the first ring-opening polymerization (ROP) of cyclic esters (lactides)5 to group transfer polymerization (GTP) of α,β-unsaturated esters (or acrylates);6 step-growth polyaddition of diols and diisocyanates to polyurethanes;7 step-growth polycondensation of diols and diesters to polyesters;8 step-growth polyaddition of dimethacrylates to unsaturated polyesters,9 which is based on fundamental steps of tail-to-tail dimerization (intermolecular umpolung) of methacrylates;10 and chain-growth conjugate polyaddition of acrylates.11 Less utilized is the basicity of the NHC for polymerization via basic activation of the protic monomer or initiator, but two recent examples invoked such activation mode to achieve oxa-Michael addition polymerization of hydroxyl functionalized acrylates to poly(ester–ether)s with Mn up to 2400 g mol−1 (ref. 12) and common (meth)acrylates in the presence of alcohol to α-alkoxy polyacrylates with Mn up to 8000 g mol−1.13 On the other hand, the phosphazene superbase, 1-tert-butyl-4,4,4-tris(dimethylamino)-2,2-bis[tris(dimethylamino) phosphoranylid-enamino]-2λ5,4λ5-catenadi(phosphazene) (tBu-P4),14 is one of the strongest known neutral bases but with low nucleophilicity. Thus, the combination of its strong basicity and low nucleophilicity renders tBu-P4 a potent organic initiator or catalyst to promote various types of polymerization reactions, but primarily as a base to deprotonate a protic initiator15 or monomer16 directly to generate anionic active species for polymerization reactions. tBu-P4 has also been reported as a nucleophilic catalyst for GTP initiated by a silyl ketene acetal.17Renewable or sustainable polymers have recently gained increasing attention, the research of which has been directed at examining the possibility of replacing petroleum-based raw materials by naturally occurring or biomass-derived renewable feedstocks for the production of polymeric materials in large commodity and specialty chemicals markets.18 In this context, renewable methylene butyrolactones based on the Tulipalin family, Tulipalin A (Fig. 1), or α-methylene-γ-butyrolactone (MBL), found in tulips,19 as well as its methyl-substituted derivatives, γ-methyl-α-methylene-γ-butyrolactone (γMMBL), derived from the biomass sourced levulinic acid,20 and β-methyl-α-methylene-γ-butyrolactone (βMMBL), derived from the biomass sourced itaconic acid,21 have been explored for the prospects of substituting the currently petroleum-based acrylic monomers, such as methyl methacrylate (MMA), in the production of acrylic bioplastics and specialty chemicals.22 Various types of polymerization processes22a have been employed to polymerize MBL, γMMBL, and βMMBL, including radical,23 anionic,24 group-transfer,25 Lewis pair zwitterionic,26 and metal-mediated coordination27 polymerization methods. NHC-mediated organopolymerization of such monomers has also been investigated.11 Regardless of the polymerization mechanism or process, all homo-polymerizations of the above three Tulipalin A-based monomers proceed exclusively through vinyl addition without ring-opening of the five-membered lactone ring.
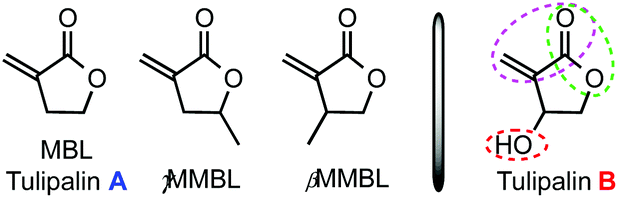 | ||
| Fig. 1 Structures of Tulipalin A, its methyl derivatives, and Tulipalin B. | ||
Tulipalin B, or β-hydroxy-α-methylene-γ-butyrolactone (βHMBL) (Fig. 1), is a biologically active (antimicrobial), naturally occurring [present as the (S)-enantiomer in tulips],19 tri-functional monomer containing an exocyclic conjugated double bond, a five-membered lactone, and a hydroxyl group. Recently, its preparation from tulip biomass by a one-step enzyme reaction has been reported.28 Perhaps owing to its structural complexity and presence of the reactive hydroxyl group, polymerization of βHMBL has not been reported in the open literature. Accordingly, the objective of this study is to investigate organopolymerization of βHMBL (1) by organic NHC and superbase catalysts.
Results and discussion
Considering the multi-functionality of the βHMBL monomer, we hypothesized that five possible scenarios could be involved in the polymerization of βHMBL by nucleophilic/basic organic NHC catalysts as outlined in Scheme 1. First, nucleophilic activation leads to a zwitterionic intermediate that could undergo repeated conjugate Michael addition11 to form vinyl addition polymer structure type I, poly(vinyl lactone) or PβHMBL, via pathway A. Second, the zwitterionic intermediate could also undergo proton transfer, with another zwitterion or monomer molecule, to generate an alkoxide species that proceeds through oxa-Michael addition29 followed by proton transfer,12 thus leading to poly(ether lactone) structure type II, via pathway B. Third, basic activation13 leads an alkoxide ion pair, which initiates the polymerization through oxa-Michael/proton transfer alternating sequences to form poly(ether lactone) structure type IIIvia pathway C. Fourth, these different pathways could interchange via pathway D, for example, after Michael addition, proton transfer could occur, which crossover to oxa-Michael addition, or vice versa, thereby forming possibly poly(vinyl-ether lactone) copolymers containing both I and II or III structure units, rather than a mixture of homopolymers. Fifth, the above possible pathways focused on the exocyclic conjugated double bond and the hydroxyl functions, but the presence of the third function, the five-membered lactone ring, points to a possible ring-opening polymerization (ROP) pathway (E), especially with a system involving alkoxide initiating/propagating species. However, the ROP of five-membered lactones, including the MBL core, is not possible (for repeated ROP enchainment) under normal conditions, due to thermodynamic reasons,30 but alkoxide species IV resulted possibly from a ring-opening event could proceed with oxa-Michael addition for further chain growth, only if this ring-opening pathway is competitive with other pathways described above.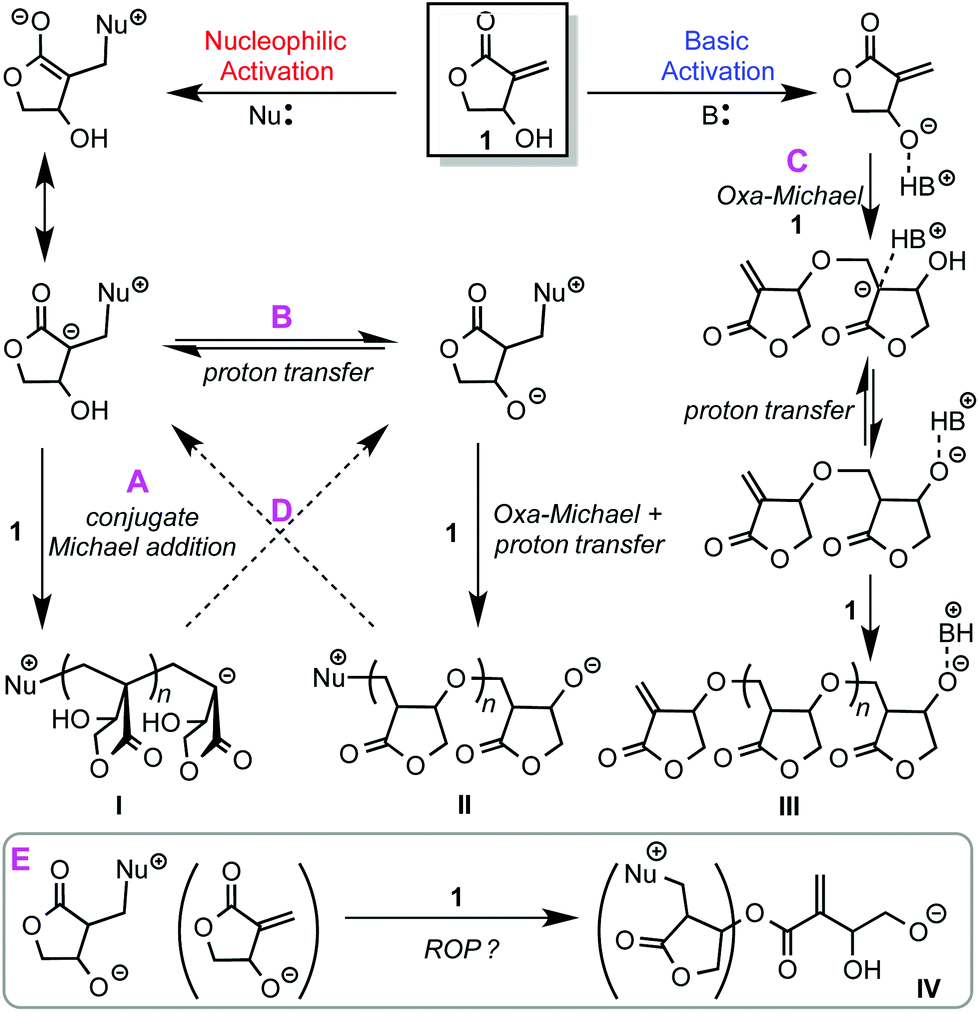 | ||
| Scheme 1 Hypothesized possible scenarios involved in the polymerization of βHMBL by nucleophilic/basic organic NHC catalysts. | ||
As outlined above, the polymerization of the tri-functional βHMBL by the nucleophilic/basic organic NHC catalysts can be very complicated. Accordingly, we chose four organic initiators/catalysts including two NHCs, ItBu (strongest nucleophile) and TPT (weaker nucleophile), one superbase, tBu-P4 (strongest base, weak nucleophile), and one radical initiator, AIBN, as a control, for examining their polymerization behavior (Table 1). For solubility reasons, DMF was chosen as the solvent. At ambient temperature (∼25 °C) in a low monomer [M] to initiator [I] ratio of 20, ItBu promoted βHMBL polymerization to completion in 24 h, producing a polymer product with Mn = 1.32 × 104 g mol−1 and Đ = 1.40 (run 1). Increasing the [M]/[I] ratio to 100, the polymerization became sluggish, achieving only 27.9% monomer conversion after 72 h (run 2). The Mn value (9.28 × 103) of the resulting polymer was also lower, due to the low monomer conversion and chain transfer (vide infra). Raising the polymerization temperature to 80 and 100 °C enabled quantitative monomer conversion in 24 h for [M]/[I] = 50, 100, 200 ratio runs or 48 h for the [M]/[I] = 500 run, but the Mn value of the resulting polymers remained in a narrow range from Mn = 7.33 × 103 g mol−1 to 8.42 × 103 g mol−1 (runs 3–6), indicating further chain-growth is limited with more monomer presumably due to chain transfer via the OH group. As compared to ItBu, the polymerization by the less nucleophilic TPT was less effective, achieving lower monomer conversions and producing polymers with lower Mn values for runs with [M]/[I] = 20 (25 °C, run 7) and 50 (80 °C, run 8), but the polymerization with [M]/[I] = 100 (100 °C) was nearly the same (run 9 vs. 4). The results of the polymerization by the superbase tBu-P4 (runs 10–14) were rather similar to those by ItBu, hinting a common polymerization mechanism for both initiators. Control runs by AIBN showed that this free radical polymerization produced PβHMBL with lower Mn and Đ values (run 15), especially when carried out in CHCl3 (Mn = 3.0 × 103 g mol−1, run 16), due to the poor solubility of the polymer in this solvent and chain transfer by this solvent, and the Mn value was not obviously affected by varying the [M]/[I] ratio from 50 to 1200. Overall, these results showed that βHMBL can be effectively polymerized into unimodal polymers with Mn up to 1.32 × 104 g mol−1, but the polymerization by the current initiating systems lack control over polymer Mn and Đ values.
| Run no. | Initiator (I) | [M]/[I] | Temp. (°C) | Time (h) | Conv.b (%) |
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (kg mol−1) c (kg mol−1) |
Đ (Mw/Mn) |
|---|---|---|---|---|---|---|---|
| a Monomer concentration [M] = 2.2 mol L−1 in DMF. b Monomer conversion measured by 1H NMR. c Number-average molecular weight (Mn) and molecular weight distribution (Đ = Mw/Mn) determined by gel-permeation chromatography (GPC) relative to poly(methyl methacrylate) standards. d Carried out in CHCl3. | |||||||
| 1 | ItBu | 20 | 25 | 24 | 100 | 13.2 | 1.40 |
| 2 | ItBu | 100 | 25 | 72 | 27.9 | 9.28 | 1.15 |
| 3 | ItBu | 50 | 80 | 24 | 100 | 7.43 | 1.50 |
| 4 | ItBu | 100 | 100 | 24 | 100 | 8.27 | 1.32 |
| 5 | ItBu | 200 | 100 | 24 | 100 | 7.33 | 1.24 |
| 6 | ItBu | 500 | 100 | 48 | >99 | 8.42 | 1.30 |
| 7 | TPT | 20 | 25 | 24 | 84.3 | 5.28 | 1.23 |
| 8 | TPT | 50 | 80 | 24 | 88.8 | 5.16 | 1.40 |
| 9 | TPT | 100 | 100 | 24 | >99 | 8.77 | 1.24 |
| 10 | t Bu-P4 | 20 | 25 | 24 | 88.4 | 8.60 | 1.23 |
| 11 | t Bu-P4 | 50 | 80 | 24 | 100 | 6.37 | 1.50 |
| 12 | t Bu-P4 | 100 | 100 | 24 | 100 | 10.2 | 1.35 |
| 13 | t Bu-P4 | 200 | 100 | 24 | 86.4 | 11.0 | 1.29 |
| 14 | t Bu-P4 | 500 | 100 | 72 | >99 | 8.26 | 1.30 |
| 15 | AIBN | 500 | 80 | 24 | 62.8 | 7.45 | 1.16 |
| 16d | AIBN | 500 | 80 | 24 | 96.0 | 3.00 | 1.10 |
Thermal properties of the resulting polymer materials were analyzed by thermal gravimetric analysis (TGA) and differential scanning calorimetry (DSC). The TGA curves (Fig. 2) showed that all the polymer materials, irrespective of the initiator used, exhibited a similar 3-step decomposition profile as well as similar initial and end onset decomposition temperatures of about 200 °C and 475 °C, respectively. However, the amount of the stable residue left at >600 °C varied significantly, from 15% by AIBN, to 22% by TPT, 29% by ItBu, and 31% by tBu-P4. DSC curves (Fig. 3) showed a similar glass-transition temperature (Tg) of −15 °C by ItBu, −17 °C by TPT, and −18 °C by tBu-P4, but no apparent Tg was observed before decomposition for the PβHMBL produced by AIBN.
 | ||
| Fig. 2 TGA curves of the polymers produced by tBu-P4 (red, run 12), ItBu (blue, run 6), TPT (green, run 9), and AIBN (black, run 15). | ||
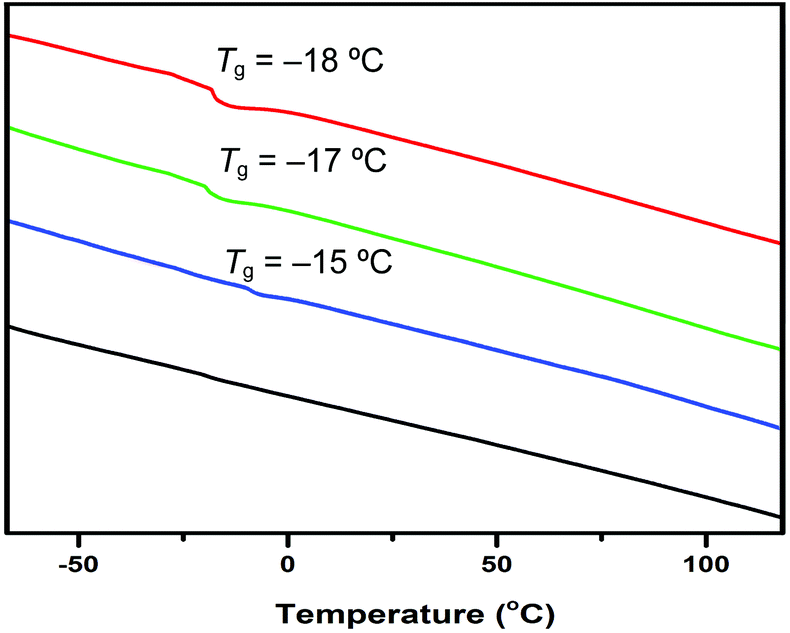 | ||
| Fig. 3 DSC traces of the polymers produced by tBu-P4 (red), TPT (green), ItBu (blue), and AIBN (black). | ||
Intriguingly, 1H NMR spectra (Fig. 4) of the polymers produced by ItBu, tBu-P4 and TPT are essentially superimposable, so are their 13C NMR spectra (Fig. S5–S7†), indicating that they all produced more or less the same polymer structure. This observation suggests that either all three organic catalysts follow the same initiation and propagation pathway or they proceed with different initiation pathways but the resulting propagating species are interchangeable thus leading to the similar polymer structure as outlined in Scheme 1. On the other hand, the PβHMBL produced by AIBN is that of a typical vinyl addition (atactic) polymer (Fig. 4), and its absence of the apparent Tg before its decomposition is consistent with this structure as the analogous polymer of βMMBL exhibited a high Tg of ∼290 °C.27 However, the polymer produced by ItBu, tBu-P4 and TPT is different than that by AIBN. The most visible spectroscopic difference is the appearance of the broad signal centered at δ 7.5 ppm in 1H NMR, which correlates to signals at δ 156–152 ppm in 13C NMR established by 1H–13C gHMQC and TOCSY (Fig. S14 and S15†), assignable to trisubstituted olefinic groups (vide infra). The presence of the OH groups in the polymer was confirmed by the H–D exchange experiment that showed the disappearance of the signal at δ 5.5 ppm upon addition of D2O (Fig. S9 and S10†) and by FT-IR that revealed a broad absorption band centered at ∼3400 cm−1 (Fig. S11†).
 | ||
| Fig. 4 Overlay of 1H NMR (DMSO-d6, 400 MHz, 25 °C) spectra of the polymers produced by TPT (green, run 9), tBu-P4 (red, run 12), ItBu (blue, run 6), and AIBN (black, run 15). | ||
Focusing on the polymer produced by ItBu, we carried out a series of 1D and 2D NMR (500 MHz) studies in DMSO-d6 at 80 °C. The 13C NMR spectrum (Fig. S5†) showed a complex pattern of resonances, but the 135-DEPT experiment (Fig. S8†) enabled initial assignments of carbon resonances as follows: δ 178–172 ppm for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, 156–152 for >CCH–, 128–126 for >CCH–, 86.8 (m) for atactic main-chain quaternary C's, 79.3 for –OCH, 75.0 and 73.3 for –OCH2, 72.1 for –OCH, 71.0 for –OCH2, 68.8 for –OCH, 44.4 for CH, and 32.1 (m) for CH2. Next, 1H–13C gHMQC and TOCSY spectra (Fig. S14 and S15†) showed correlation between the peak at δ 7.5 ppm in 1H NMR and the peaks at δ 156–152 ppm in 13C NMR, confirming the assignment of the peak at 7.5 ppm for >CCH–. These correlation spectra also enabled assignments of the resonances from δ 2.7–2.0 ppm in 1H NMR for those of CH (lower field) and CH2 (higher field) protons, as well as the peak at δ 4.8 ppm for –OCH2 protons and δ 4.4–3.9 ppm for those of –OCH and additional –OCH2 protons (Fig. 5). In addition, from the 1H–1H gCOSY spectrum (Fig. S12†), the proton in >CCH– at δ 7.5 ppm correlates to the protons in –OCH2 at δ 4.8 ppm, thus revealing a connectivity of >CCH–CH2O–. Furthermore, the 1H–1H zTOCSY spectra (Fig. S13†) revealed that both protons in >CCH–CH2O– correlate to CH2 protons centered at about δ 2.6 ppm, and the OH proton correlates to both types of the methylene protons in –OCH2 and CH2, revealing the intact hydroxyl lactone motif in the vinyl addition polymer units. Putting all pieces together, the possible structures present in the resulting polymer contain not only poly(vinyl lactone) (type I) and poly(ether lactone) (type II or III) but also a structure containing the trisubstituted olefinic moiety >CCH–CH2O–, the origin of which is probably due to dehydration (vide infra).
O, 156–152 for >CCH–, 128–126 for >CCH–, 86.8 (m) for atactic main-chain quaternary C's, 79.3 for –OCH, 75.0 and 73.3 for –OCH2, 72.1 for –OCH, 71.0 for –OCH2, 68.8 for –OCH, 44.4 for CH, and 32.1 (m) for CH2. Next, 1H–13C gHMQC and TOCSY spectra (Fig. S14 and S15†) showed correlation between the peak at δ 7.5 ppm in 1H NMR and the peaks at δ 156–152 ppm in 13C NMR, confirming the assignment of the peak at 7.5 ppm for >CCH–. These correlation spectra also enabled assignments of the resonances from δ 2.7–2.0 ppm in 1H NMR for those of CH (lower field) and CH2 (higher field) protons, as well as the peak at δ 4.8 ppm for –OCH2 protons and δ 4.4–3.9 ppm for those of –OCH and additional –OCH2 protons (Fig. 5). In addition, from the 1H–1H gCOSY spectrum (Fig. S12†), the proton in >CCH– at δ 7.5 ppm correlates to the protons in –OCH2 at δ 4.8 ppm, thus revealing a connectivity of >CCH–CH2O–. Furthermore, the 1H–1H zTOCSY spectra (Fig. S13†) revealed that both protons in >CCH–CH2O– correlate to CH2 protons centered at about δ 2.6 ppm, and the OH proton correlates to both types of the methylene protons in –OCH2 and CH2, revealing the intact hydroxyl lactone motif in the vinyl addition polymer units. Putting all pieces together, the possible structures present in the resulting polymer contain not only poly(vinyl lactone) (type I) and poly(ether lactone) (type II or III) but also a structure containing the trisubstituted olefinic moiety >CCH–CH2O–, the origin of which is probably due to dehydration (vide infra).
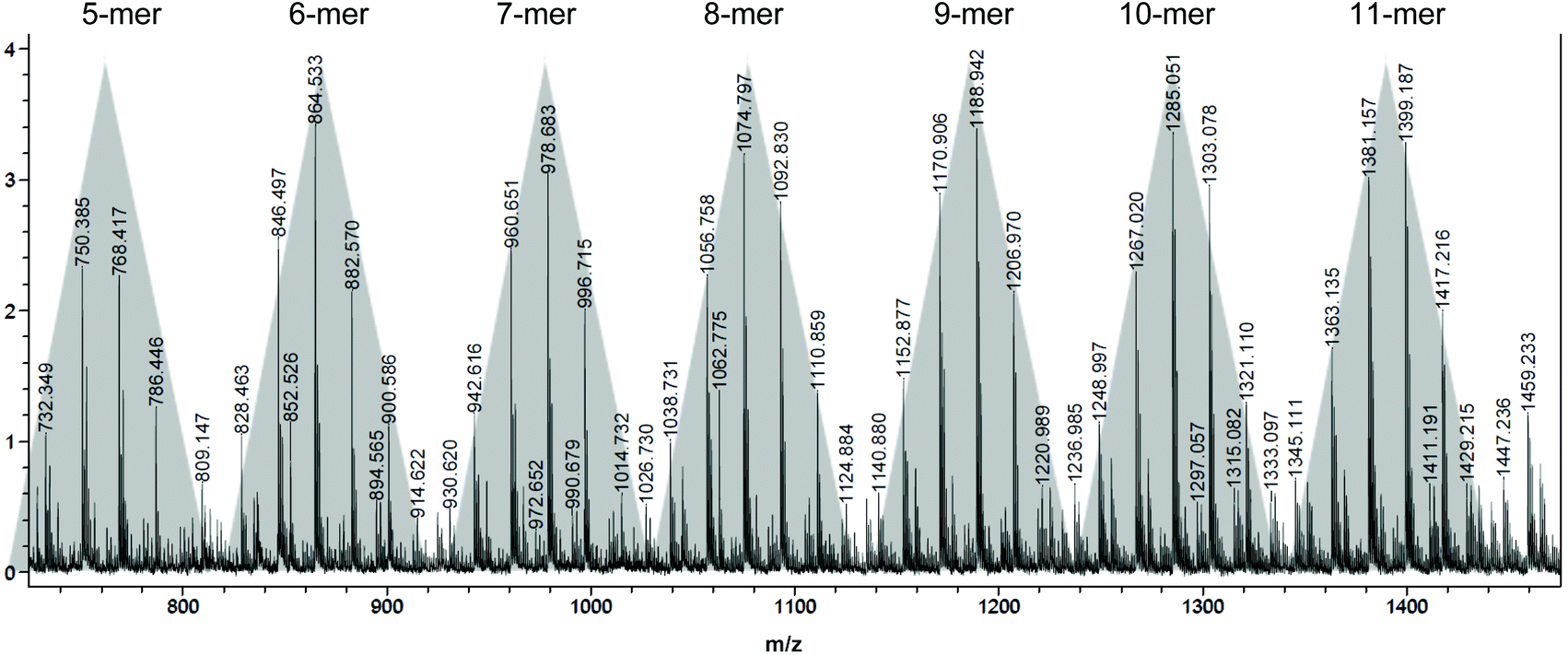 | ||
| Fig. 5 MALDI-TOF MS spectrum of the low molecular weight oligomers produced by ItBu. | ||
The question of whether these structures represent a mixture of different polymers or possibly a copolymer containing all such units was interrogated by solvent fractionation experiments. We reasoned that polyether homopolymers would be soluble in solvents such as chloroform;31 accordingly, the crude polymer was subjected to Soxhlet extraction in refluxing chloroform for 30 h. There was no soluble fraction extracted, suggesting that the polymer product was not a mixture of different polymers, which is consistent with the unimodal molecular weight distribution on their GPC traces. Another key question concerns the >CCH–CH2O– unit derived from the above spectroscopic analysis and how it is generated in the current polymerization system. In principle, dehydration of an oxa-Michael addition product (see Scheme 1) could lead to the α,β-unsaturated-γ-butyrolactone unit, but its mechanistic pathway is currently unclear. Furthermore, the OH group on the vinyl polymer units could participate in proton transfer and subsequently generate a branching structure via the oxa-Michael addition pathway. In addition, the appearance of several types of >CCH– signals at δ 156–152 ppm in 13C NMR may indicate that dehydration could occur within the vinyl polymer repeat unit to generate more such unsaturated structure. The observed low Tg of −15 °C for the polymer produced by ItBu is also consistent with a copolymer containing the soft (branched) polyether units.
MALDI-TOF mass spectrum (Fig. 5) of a low molecular weight polymer sample produced by ItBu showed a series of peaks associated with different sets of isomerides, with the peaks in the same shade-highlighted set representing the same total degrees of copolymerization but with different degrees of dehydration (loss of the mass unit of 18). Therefore, for the polymer produced by ItBu the masses m/z can be expressed as: m/z = Mend + (114.1–18.0) × m + 114.1 × n, where m and n are the degrees of polymerization for βHMBL with dehydration and the degrees of polymerization for βHMBL without dehydration, respectively, while Mend is the MW of chain ends. Take the last set as an example, the peaks in this set can be attributed to the 11th-mer poly(vinyl-ether lactone) copolymer, where the peaks at 1417.216, 1399.187, 1381.157, and 1363.135 represent the copolymers containing 11 units of βHMBL with 2 water loss, 11 units of βHMBL with 3 water loss, 11 units of βHMBL with 4 water loss, and 11 units of βHMBL with 5 water loss, respectively. According to this analysis, the MW of the chain end is calculated to be 179.66 g mol−1 (Fig. 6), corresponding to ItBu+.
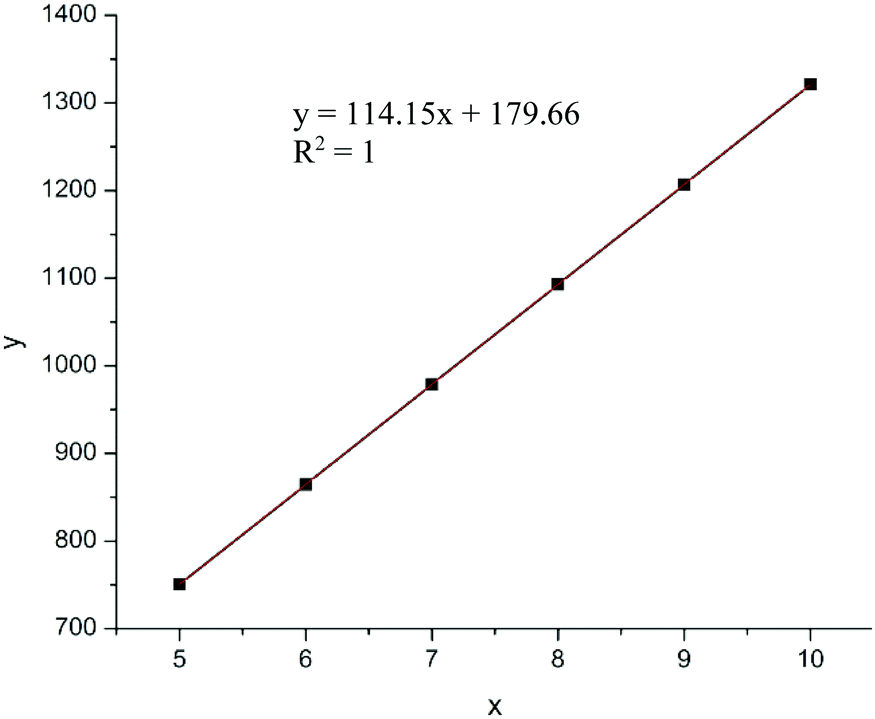 | ||
| Fig. 6 Plot of the m/z value of the molecular ions (y) vs.βHMBL repeat unit (x). | ||
Conclusions
In summary, we have revealed for the first time the successful polymerization of the naturally occurring, tri-functional monomer Tulipalin B or βHMBL using nucleophilic/basic organic catalysts and a radical initiator, producing polymer products with molecular weight up to Mn = 1.32 × 104 g mol−1. The polymer produced by AIBN is that of a typical vinyl addition (atactic) polymer, but the polymers produced by NHCs ItBu and TPT as well as superbase tBu-P4 have rather complex structures, although the structures by ItBu, TPT, tBu-P4 are essentially the same. Extensive spectroscopic and analytical analyses of the resulting polymer products led to a possible conclusion that the polymer is likely a branched copolymer of poly(vinyl-ether lactone)s, derived from the proposed mechanistic crossovers between conjugate Michael and oxa-Michael additions, enabled by proton transfer due to the presence of OH groups in the monomer and within the polymer chain, followed by dehydration and branching.Acknowledgements
This work was supported by the US National Science Foundation (NSF-1300267).Notes and references
- Selected recent reviews: (a) C. Thomas and B. Bibal, Green Chem., 2014, 16, 1687–1699 RSC; (b) H. A. Brown and R. M. Waymouth, Acc. Chem. Res., 2013, 46, 2585–2596 CrossRef CAS PubMed; (c) K. Fuchise, Y. Chen, T. Satoh and T. Kakuchi, Polym. Chem., 2013, 4, 4278–4291 RSC; (d) H. A. Brown and R. M. Waymouth, Acc. Chem. Res., 2013, 46, 2585–2596 CrossRef CAS PubMed; (e) M. K. Kiesewetter, E. J. Shin, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2010, 43, 2093–2107 CrossRef CAS; (f) N. E. Kamber, W. Jeong, R. M. Waymouth, R. C. Pratt, B. G. G. Lohmeijer and J. L. Hedrick, Chem. Rev., 2007, 107, 5813–5840 CrossRef CAS PubMed.
- Selected recent reviews: (a) C. M. R. Volla, I. Atodiresei and M. Rueping, Chem. Rev., 2014, 114, 2390–2431 CrossRef CAS PubMed; (b) D. Liu and E. Y.-X. Chen, Green Chem., 2014, 16, 964–981 RSC; (c) R. C. Wende and P. Schreiner, Green Chem., 2012, 14, 1821–1849 RSC; (d) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167–178 CrossRef CAS PubMed; (e) T. Marcelli and H. Hiemstra, Synthesis, 2010, 1229–1279 CrossRef CAS; (f) D. W. C. MacMillan, Nature, 2008, 455, 304–308 CrossRef CAS PubMed.
- Selected recent reviews: (a) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed; (b) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed; (c) D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC; (d) S. J. Ryan, L. Candish and D. W. Lupton, Chem. Soc. Rev., 2013, 42, 4906–4917 RSC; (e) A. Grossmann and D. Enders, Angew. Chem., Int. Ed., 2012, 51, 314–325 CrossRef CAS PubMed; (f) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC; (g) T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 50, 6940–6952 CrossRef PubMed; (h) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed; (i) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed; (j) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed; (k) N. Marion, S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988–3000 CrossRef CAS PubMed; (l) D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–91 CrossRef CAS PubMed.
- Selected recent reviews: (a) S. Naumann and A. P. Dove, Polym. Chem., 2015, 6, 3185–3200 RSC; (b) M. Fèvre, J. Pinaud, Y. Gnanou, J. Vignolle and D. Taton, Chem. Soc. Rev., 2013, 42, 2142–2172 RSC.
- E. F. Connor, G. W. Nyce, M. Myers, A. Möck and J. L. Hedrick, J. Am. Chem. Soc., 2002, 124, 914–915 CrossRef CAS PubMed.
- (a) K. Fuchise, Y. Chen, T. Satoh and T. Kakuchi, Polym. Chem., 2013, 4, 4278–4291 RSC; (b) J. Raynaud, A. Ciolino, A. Baceiredo, M. Destarac, F. Bonnette, T. Kato, Y. Gnanou and D. Taton, Angew. Chem., Int. Ed., 2008, 47, 5390–5393 CrossRef CAS PubMed; (c) M. D. Scholten, J. L. Hedrick and R. M. Waymouth, Macromolecules, 2008, 41, 7399–7404 CrossRef CAS.
- O. Coutelier, M. El Ezzi, M. Destarac, F. Bonnette, T. Kato, A. Baceiredo, G. Sivasankarapillai, Y. Gnanou and D. Taton, Polym. Chem., 2012, 3, 605–608 RSC.
- G. W. Nyce, J. A. Lamboy, E. F. Connor, R. M. Waymouth and J. L. Hedrick, Org. Lett., 2002, 4, 3587–3590 CrossRef CAS PubMed.
- M. Hong and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2014, 53, 11900–11906 CrossRef CAS PubMed.
- (a) T. Kato, Y. Ota, S.-I. Matsuoka, K. Takagi and M. Suzuki, J. Org. Chem., 2013, 78, 8739–8747 CrossRef CAS PubMed; (b) S.-I. Matsuoka, Y. Ota, A. Washio, A. Katada, K. Ichioka, K. Takagi and M. Suzuki, Org. Lett., 2011, 13, 3722–3725 CrossRef CAS PubMed; (c) A. T. Biju, M. Padmanaban, N. E. Wurz and F. Glorius, Angew. Chem., Int. Ed., 2011, 50, 8412–8415 CrossRef CAS PubMed.
- (a) Y. Zhang, M. Schmitt, L. Falivene, L. Caporaso, L. Cavallo and E. Y.-X. Chen, J. Am. Chem. Soc., 2013, 135, 17925–17942 CrossRef CAS PubMed; (b) Y. Zhang and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2012, 51, 2465–2469 CrossRef CAS PubMed.
- S.-I. Matsuoka, S. Namera and M. Suzuki, Polym. Chem., 2015, 6, 294–301 RSC.
- W. N. Ottou, D. Bourichon, J. Vignolle, A.-L. Wirotius, F. Robert, Y. Landais, J.-M. Sotiropoulos, K. Miqueu and D. Taton, Chem. – Eur. J., 2015, 21, 9447–9453 CrossRef CAS PubMed.
- (a) I. Leito, T. Rodima, I. A. Koppel, R. Schwesinger and V. M. Vlasov, J. Org. Chem., 1997, 62, 8479–8483 CrossRef CAS PubMed; (b) R. Schwesinger, C. Hasenfratz, H. Schlemper, L. Walz, E. M. Peters, K. Peters and H. G. von Schnering, Angew. Chem., Int. Ed. Engl., 1993, 32, 1361–1363 CrossRef; (c) R. Schwesinger and H. Schlemper, Angew. Chem., Int. Ed. Engl., 1987, 26, 1167–1169 CrossRef; (d) R. Schwesinger, Chimia, 1985, 39, 269–272 CAS.
- (a) J. Zhao, N. Hadjichristidis and Y. Gnanou, Polimery, 2014, 59, 49–59 CrossRef CAS; (b) B. Esswein and M. Möller, Angew. Chem., Int. Ed. Engl., 1996, 35, 623–625 CrossRef CAS; (c) B. Eßwein, N. M. Steidl and M. Möller, Macromol. Rapid Commun., 1996, 17, 143–148 CrossRef; (d) A. Molenberg and M. Möller, Macromol. Rapid Commun., 1995, 16, 449–453 CrossRef CAS; (e) T. Pietzonka and D. Seebach, Angew. Chem., Int. Ed. Engl., 1993, 32, 716–717 CrossRef.
- M. Schmitt, L. Falivene, L. Caporaso, L. Cavallo and E. Y.-X. Chen, Polym. Chem., 2014, 5, 3261–3270 RSC.
- (a) Y. Chen, K. Fuchise, A. Narumi, S. Kawaguchi, T. Satoh and T. Kakuchi, Macromolecules, 2011, 44, 9091–9098 CrossRef CAS; (b) J.-C. Hsu, Y. Chen, T. Kakuchi and W.-C. Chen, Macromolecules, 2011, 44, 5168–5177 CrossRef CAS; (c) T. Kakuchi, Y. Chen, J. Kitakado, K. Mori, K. Fuchise and T. Satoh, Macromolecules, 2011, 44, 4641–4647 CrossRef CAS.
- Selected recent reviews: (a) M. J.-L. Tschan, E. Brulé, P. Haquette and C. M. Thomas, Polym. Chem., 2012, 3, 836–851 RSC; (b) G. W. Coates and M. A. Hillmyer, A virtual issue on “Polymers from Renewable Resources”, Macromolecules, 2009, 42, 7987–7989 CrossRef CAS; (c) A. Gandini, Macromolecules, 2008, 41, 9491–9504 CrossRef CAS; (d) C. K. Wiliams and M. A. Hillmyer, Polym. Rev., 2008, 48, 1–10 CrossRef; (e) M. A. R. Meier, J. O. Metzger and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1788–1802 RSC.
- (a) R. R. A. Kitson, A. Millemaggi and R. J. K. Taylor, Angew. Chem., Int. Ed., 2009, 48, 9426–9451 CrossRef CAS PubMed; (b) H. M. R. Hoffman and J. Rabe, Angew. Chem., Int. Ed. Engl., 1985, 24, 94–110 CrossRef.
- (a) L. E. Manzer, ACS Symp. Ser., 2006, 921, 40–51 CrossRef CAS; (b) L. E. Manzer, Appl. Catal., A, 2004, 272, 249–256 CrossRef CAS.
- R. R. Gowda and E. Y.-X. Chen, Org. Chem. Front., 2014, 1, 230–234 RSC.
- Recent reviews: (a) R. R. Gowda and E. Y.-X. Chen, in Encyclopedia of Polymer Science and Technology, ed. H. F. Mark, Wiley, Hoboken, 4th edn, 2013, vol. 8, pp. 235–271, 2014 (print edition), DOI:10.1002/0471440264.pst606 (online edition); (b) S. Agarwal, Q. Jin and S. Maji, ACS Symp. Ser., 2012, 1105, 197–212 CrossRef CAS; (c) R. Mullin, Chem. Eng. News, 2004, 82(45), 29–37 CrossRef.
- Selected recent examples: (a) Y. Higaki, R. Okazaki and A. Takahara, ACS Macro Lett., 2012, 1, 1124–1127 CrossRef CAS; (b) R. A. Cockburn, R. Siegmann, K. A. Payne, S. Beuermann, T. F. L. McKenna and R. A. Hutchinson, Biomacromolecules, 2011, 12, 2319–2326 CrossRef CAS PubMed; (c) R. A. Cockburn, T. F. L. McKenna and R. A. Hutchinson, Macromol. Chem. Phys., 2010, 211, 501–509 CrossRef CAS; (d) J. Mosnáček, J. A. Yoon, A. Juhari, K. Koynov and K. Matyjaszewski, Polymer, 2009, 50, 2087–2094 CrossRef; (e) J. Mosnáček and K. Matyjaszewski, Macromolecules, 2008, 41, 5509–5511 CrossRef; (f) G. Qi, M. Nolan, F. J. Schork and C. W. Jones, J. Polym. Sci., Polym. Chem., 2008, 46, 5929–5944 CrossRef CAS; (g) C. U. Pittman Jr. and H. Lee, J. Polym. Sci., Polym. Chem., 2003, 41, 1759–1777 CrossRef; (h) J. W. Stansbury and J. M. Antonucci, Dent. Mater., 1992, 8, 270–273 CrossRef CAS PubMed; (i) M. Ueda, M. Takahashi, Y. Imai and C. U. Pittman Jr., J. Polym. Sci., Polym. Chem. Ed. Engl., 1982, 20, 2819–2828 CrossRef CAS; (j) M. K. Akkapeddi, Polymer, 1979, 20, 1215–1216 CrossRef CAS.
- (a) Y. Hu, L. O. Gustafson, H. Zhu and E. Y.-X. Chen, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2008–2017 CrossRef CAS; (b) J. Suenaga, D. M. Sutherlin and J. K. Stille, Macromolecules, 1984, 17, 2913–2916 CrossRef CAS; (c) M. K. Akkapeddi, Macromolecules, 1979, 12, 546–551 CrossRef CAS.
- (a) Y. Zhang, L. O. Gustafson and E. Y.-X. Chen, J. Am. Chem. Soc., 2011, 133, 13674–13684 CrossRef CAS PubMed; (b) G. M. Miyake, Y. Zhang and E. Y.-X. Chen, Macromolecules, 2010, 43, 4902–4908 CrossRef CAS; (c) D. Y. Sogah, W. R. Hertler, O. W. Webster and G. M. Cohen, Macromolecules, 1987, 20, 1473–1488 CrossRef CAS.
- (a) T. Xu and E. Y.-X. Chen, J. Am. Chem. Soc., 2014, 136, 1774–1777 CrossRef CAS PubMed; (b) Y. Zhang, G. M. Miyake, M. G. John, L. Falivene, L. Caporaso, L. Cavallo and E. Y.-X. Chen, Dalton Trans., 2012, 41, 9119–9134 RSC; (c) Y. Zhang, G. M. Miyake and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2010, 49, 10158–10162 CrossRef CAS PubMed.
- (a) R. R. Gowda and E. Y.-X. Chen, Dalton Trans., 2013, 42, 9263–9273 RSC; (b) Y. Hu, X. Wang, Y. Chen, L. Caporaso, L. Cavallo and E. Y.-X. Chen, Organometallics, 2013, 32, 1459–1465 CrossRef CAS; (c) X. Chen, L. Caporaso, L. Cavallo and E. Y.-X. Chen, J. Am. Chem. Soc., 2012, 134, 7278–7281 CrossRef CAS PubMed; (d) Y. Hu, G. M. Miyake, B. Wang, D. Cui and E. Y.-X. Chen, Chem. – Eur. J., 2012, 18, 3345–3354 CrossRef CAS PubMed; (e) Y. Hu, X. Xu, Y. Zhang, Y. Chen and E. Y.-X. Chen, Macromolecules, 2010, 43, 9328–9336 CrossRef CAS; (f) G. M. Miyake, S. E. Newton, W. R. Mariott and E. Y.-X. Chen, Dalton Trans., 2010, 39, 6710–6718 RSC.
- T. Nomura, E. Hayashi, S. Kawakami, S. Ogita and Y. Kato, Biosci., Biotechnol., Biochem., 2015, 79, 25–35 CrossRef CAS PubMed.
- E. M. Phillips, M. Riedrich and K. A. Scheidt, J. Am. Chem. Soc., 2010, 132, 13179–13181 CrossRef CAS PubMed.
- (a) M. Hong and E. Y.-X. Chen, Macromolecules, 2014, 47, 3614–3624 CrossRef CAS; (b) A. Duda and A. Kowalski, Thermodynamics and kinetics of ring-opening polymerization, in Handbook of Ring-Opening Polymerization, ed. P. Dubois, O. Coulembier and J.-M. Raquez, Wiley-VCH, Weinheim, 2009, ch. 3 Search PubMed.
- (a) T. Wang, M. Yan, X. Sun and D. Quan, Polymer, 2015, 57, 21–28 CrossRef CAS; (b) D. Wang, G. Zhang, Y. Zhang, Y. Gao, Y. Zhao, C. Zhou, Q. Zhang and X. Wang, J. Appl. Polym. Sci., 2007, 103, 417–424 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterisations. See DOI: 10.1039/c5qo00262a |
| This journal is © the Partner Organisations 2015 |