
Autoinductive thiolation/oxygenation of alkenes at room temperature†
Qingquan
Lu
a,
Huamin
Wang
a,
Pan
Peng
a,
Chao
Liu
a,
Zhiyuan
Huang
a,
Yi
Luo
a and
Aiwen
Lei
*ab
aCollege of Chemistry and Molecular Sciences, the Institute for Advanced Studies (IAS), Wuhan University, Wuhan, 430072, P. R. China. E-mail: aiwenlei@whu.edu.cn; Fax: +(86) 27-68754672
bNational Research Center for Carbohydrate Synthesis, Jiangxi Normal University, Nanchang 330022, Jiangxi, P. R. China
First published on 7th May 2015
Abstract
A new reaction for O2 fixation is rationally demonstrated, in which radical thiolation/oxygenation of activated alkenes proceeds spontaneously at room temperature with no need for any additives. EPR and operando IR experiments revealed that O2 serves as the initiator to trigger this radical reaction and that autoinductive processes are the critical factor to drive the reaction.
Dioxygen transformation has great usage in synthetic chemistry, bioinorganic chemistry, enzymology etc., and the development of selective oxygenation reactions is a fundamentally key issue in modern organic synthesis.1 Consequently, consistently significant effort has been devoted to dioxygen activation, and numerous methodologies relying on enzymes, transition metal complexes etc. have been established,2 whereas spontaneous dioxygen fixation under benign conditions is quite limited.1–3 Furthermore, the necessity of multifarious activators in these processes is a long-standing challenge to be addressed, which often results in affording a mixture of products.1–3 Thus, the exploration of green and new strategies to update these methods is pressingly worthwhile.
As a result of their friendly, sustainable and economic advantages, dioxygen-triggered radical reactions have received much attention, which could serve as an ideal and promising route to address the above-mentioned challenge.3 On this vein, aerobic ‘oxygenase-type’ difunctionalization of alkenes and oxygenation of carbonyl compounds, taking advantage of the vigorous reactivity of the substrates, have been developed recently,4,5 in which dioxygen was utilized as the mediator and multifarious alcohols could be typically furnished in the presence of suitable additives. Despite these advances, as far as we know, solely dioxygen-mediated radical thiolation/oxygenation of alkenes to efficiently prepare carbonyl compounds still remains unexplored. Herein, we describe a convenient and spontaneous thiolation/oxygenation of alkenes without the need for any redox-active catalysts and external additives, and various valuable β-keto sulfides could be synthesized by just mixing the reactants in a solvent (Scheme 1).
 | ||
| Scheme 1 Spontaneous thiolation/oxygenation of alkenes. | ||
In 2010, Stephen Caddick and coworkers have described a highly selective and reagent-free hydroacylation of α,β-unsaturated esters by using the aldehyde auto-oxidation process.6 The key to the success of this reaction lies in the electrophilic characteristic of the radical intermediate, generated from the addition of an acyl radical to the electron-poor alkenes, which is effective for H-abstraction. In principle, the reactivity of the free radical depends on the energy level of the Singly Occupied Molecular Orbital (SOMO), and the introduction of a heteroatom to a radical adjacent chain improves the electron density and stability of the free radical through the P–Pπ orbital conjugate effect, which makes the free radical more nucleophilic and easier for oxygenation.7 This provides us the oppotunity to switch the chemical selectivity in radical chemistry and achieve the preferred oxygenation process through reasonable substrate design.
According to the above point of view, we commenced our investigation by reacting phenylethylene with p-toluenethiol (2a) to test our proposal. Not surprisingly, a mixture formed from H-abstraction and oxygenation processes was observed with low yields and low selectivity. This result prompted us to select α-bromostyrene (1a) instead of phenylethylene based on our initial assumption. To our delight, 1-phenyl-2-(p-tolylthio)ethan-1-one (3aa) could be obtained at room temperature by just mixing the reactants in a solvent (for details, see Table S1†). Moreover, this reaction displayed high selectivity as we expected and no ‘thiol–ene coupling’ product or further oxidized products including sulfoxides and sulfones were observed,8 which was usually difficult to be dodged under oxidative conditions. Further experiments have shown that the solvent effect was extremely obvious in promoting the efficiency of this transformation. Low-polar solvents including CH2Cl2, MeCN and toluene gave inferior results compared with the polar ones, and the yield dramatically increased to 89% when DMF was used. Notably, 93% yield of 3aa was obtained when the reaction was carried out under O2 while no expected product was detected under anaerobic conditions. The final screening of the reaction concentration elucidated that a condensed solution is beneficial for the reaction efficiency which could furnish the best yield of 93% under air.
With the optimized conditions in hand, the generality of this reaction was next evaluated, and Scheme 2 summarizes the results. Benzenethiols bearing a wide range of either electron-donating (Me, MeO) or electron-withdrawing (Br, F, Cl, CF3) functional groups were all applicable to this protocol well, which could be smoothly transferred into the expected β-keto sulfides in good yields. Gratifyingly, ortho-methoxyl and fluoro substituted benzenethiols, which have steric hindrance at the substituents, were demonstrated to marginally affect the efficiency of this reaction by formation of the expected products (3ad and 3ai) in 86% and 75% yields. It's noteworthy to mention that the halo substituents including Br, F, Cl were proved to be intact after the reaction and provided the relevant products in good yields, which offers ample opportunities for further modifications (3ag–3ak). Moreover, an electron-rich heterocycle which may potentially be sensitive to oxidative conditions, as exemplified by thiophenethiol, was also examined under the optimal conditions, providing the product 3am in moderate yield. In addition, a series of mono- and multisubstituted α-bromostyrene derivatives was further subjected to the standard conditions. Functional groups such as thienyl, phenyl, methoxyl, naphthyl substituted on the aryl ring of the α-bromostyrene were well tolerated, delivering the corresponding products in excellent yields (3ba–3ea). Intriguingly, the sterically hindered non-terminal and cyclic α-bromostyrene derivatives were also effective in this transformation, offering the corresponding products in excellent yields (3fa and 3ga).9
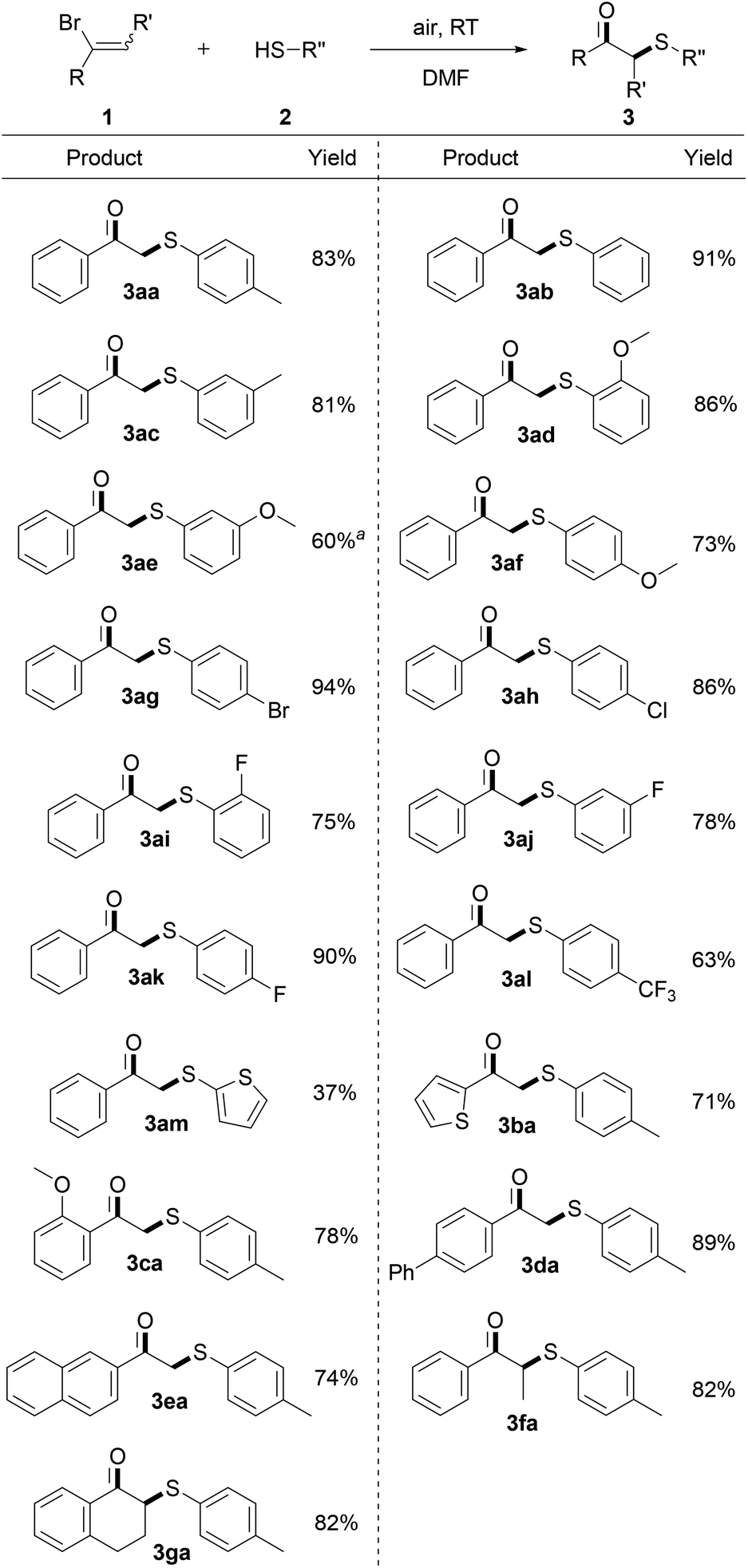 | ||
| Scheme 2 Unless otherwise specified, all reactions were carried out using 1 (0.2 mmol), 2 (0.6 mmol), in DMF (2.0 mL) under air at RT for 1 h, isolated yield. a3-Methoxythiophenol (0.8 mmol) was employed for 2 h. | ||
Proverbially, C–Cl bonds are more difficult to be cleaved because of the stronger bond strength than the C–Br bond. Inspired by the encouraging results, we next attempted to apply this protocol to a set of α-chlorostyrene derivatives and accomplish the C(sp2)–Cl bond functionalization. In a surprise twist, a variety of α-chlorostyrene derivatives, containing either electron-donating or electron-withdrawing substituents, were amenable to this reaction system as well, giving the desired products in good yields at room temperature (Scheme 3).
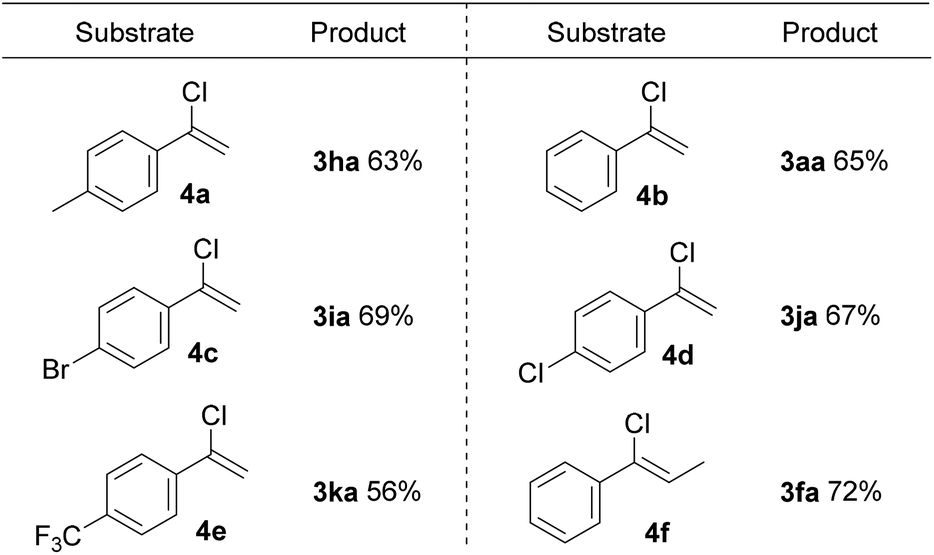 | ||
| Scheme 3 Substrate scope of α-chlorostyrene derivatives. All reactions were carried out using 4 (0.2 mmol), 2a (0.8 mmol), in DMF (2.0 mL) under air at room temperature for 1 h, isolated yield. | ||
Regarding the weak S–H bond dissociation energies (e.g., 79 kcal mol−1 for thiophenol) and the stability of the resulting arenethiyl radical, thiophenols have been extensively studied in the presence of ultraviolet light, peroxide initiators, hyperbaric oxygen etc.8 However, there have been surprisingly scarce examples towards the utilization of thiophenols in the absence of additional activators, and the detailed kinetic investigation is less as well. To gain insights into the reaction mechanism, 18O isotope labeling experiment was firstly conducted to elucidate the origination of the carbonyl oxygen atom of the β-keto sulfides (Scheme 4). The reaction between 1a and 2a under 18O2 afforded the 18O-labeled product 3aa′ in 80% yield with 60% isotopic purity. The moderate labeling ratio was likely due to the oxygen-exchange between 3aa′ and a trace amount of water in reagent under acid conditions (HBr was generated during the reaction), and H218O labeling experiment was performed further to prove this. These results demonstrated that O2 took part into this reaction and transformed into the final product.
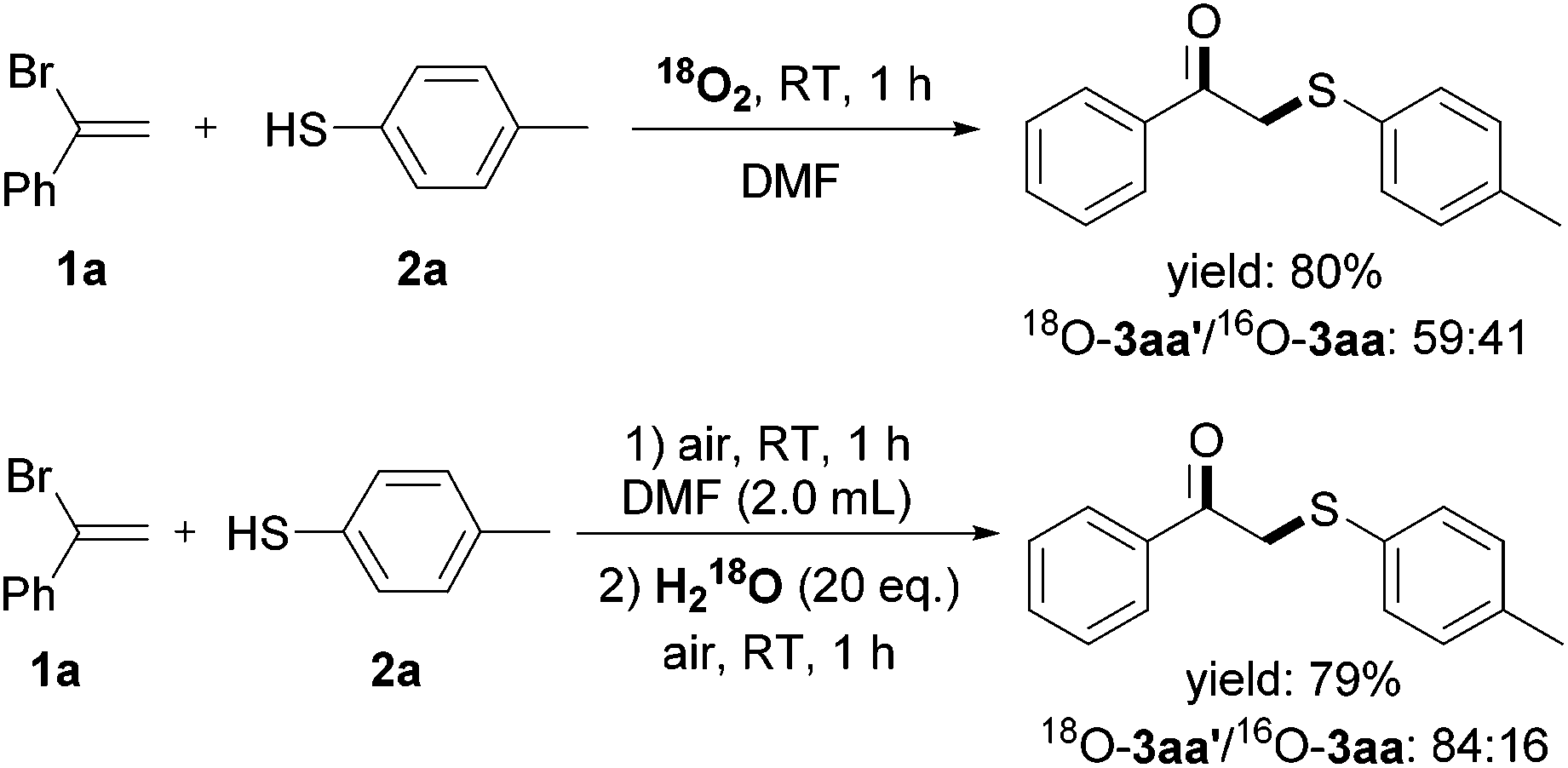 | ||
| Scheme 4 18O isotope labeling experiments. | ||
Toward a further understanding of the role of O2 in this reaction, electron paramagnetic resonance (EPR) experiments were conducted, and the results are illustrated in Fig. 1. A distinct EPR signal was detected using a DMPO (5,5-dimethyl-1-pyrroline N-oxide) spin trap when 2a was subjected to DMF under air.10 The hyperfine coupling constants are αN = 12.3 G and αH = 14.3 G. This signal was assigned to a DMPO-p-toluene thiyl radical adduct by comparison with a synthesized sample, which indicated that O2 also serves as the initiator to oxidize thiophenols to offer the corresponding S-centered radicals under the present protocol.
 | ||
| Fig. 1 (A) DMPO spin trapping of radicals formed from (red line) 2a (0.3 mmol) in DMF (2.0 mL) at RT under air, (blue line) 1a (0.1 mmol) in DMF (2.0 mL) at RT under air. (B) Comparison of DMPO-radical adduct formed in situ with DMPO-p-toluene thiyl radical adduct formed from 2a (0.3 mmol) and AIBN (0.1 mmol) in DMF (2.0 mL) at 70 °C under N2. | ||
Theoretically, all of the hydrocarbon compounds could be easily autoxidized by O2, furnishing water and CO2 as the products on the basis of thermodynamics. But the truth is that, most of the autoxidation reactions are kinetically inert in the absence of external activators. To investigate the oxidation rate of 2a, operando IR was employed to monitor the interaction between 1a/2a and air. No obvious consumption of 1a or 2a was observed (Fig. 2), suggesting that 1a and 2a were comparatively stable at this stage and the direct initiation of thiophenols by air to produce the S-centered radicals might be a slow process.
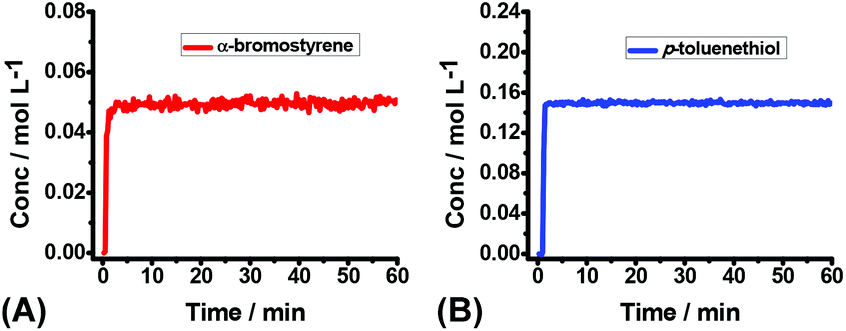 | ||
| Fig. 2 (A) The kinetic profile of the reaction of 1a (0.2 mmol) in THF (4.0 mL) at RT for 1 h under air. (B) The kinetic profile of the reaction of 2a (0.6 mmol) in THF (4.0 mL) at RT for 1 h under air. | ||
To shed more insight into the reaction mechanism, the model reaction between 1a and 2a in the presence or absence of dioxygen were also investigated by operando IR. Clearly, no reaction proceeded at all under argon (Fig. 3A), which excluded a direct interaction between 1a and 2a. Conversely, the quick consumption of both 1a and 2a was observed as the desired product 3aa increased concurrently under air (Fig. 3B). Combining the available information in operando IR experiments, we may draw a conclusion that molecular oxygen serves as the initiator but not the dominator to trigger the reaction, and the autoinductive process was involved in this transformation, which reinitiated radical chain processes and dominated reaction rates.
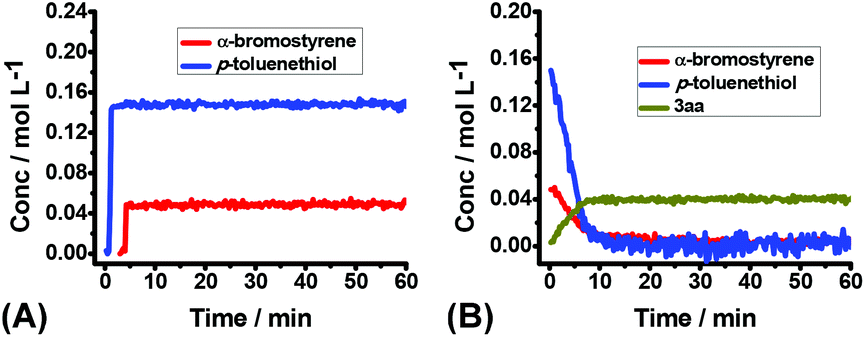 | ||
| Fig. 3 The kinetic profile of the reaction of 1a (0.2 mmol) and 2a (0.6 mmol) in THF (4.0 mL) at RT for 1 h: (A) under argon; (B) under air. | ||
In addition, a considerable amount of aryl disulfide was detected after the reaction (for details, see the ESI†), which could serve as another evidence to support that the autoinductive process dominated this transformation.11 This result also disclosed that thiophenols may not only act as a substrate to participate the reaction, but also as a reductant quenching the oxidant generated in situ.12
Although the detailed mechanism remains to be elucidated, a tentative reaction pathway is proposed (Scheme 5).13 Benzenethiol is firstly oxidized by dioxygen to release arenethiyl radical I, and this step might be a slow process. Then a carbon-centered radical II could be generated by means of radical addition of I to α-bromostyrene (1a), which can be trapped by O2 to afford peroxyl radical III. Subsequently, the hydroperoxide intermediate IV is furnished through an intermolecular hydrogen abstraction with the release of another arenethiyl radical I. Finally, intermediate IV undergoes a reduction process and a subsequent nucleophilic substitution step would afford the target molecule 3ab.14,15
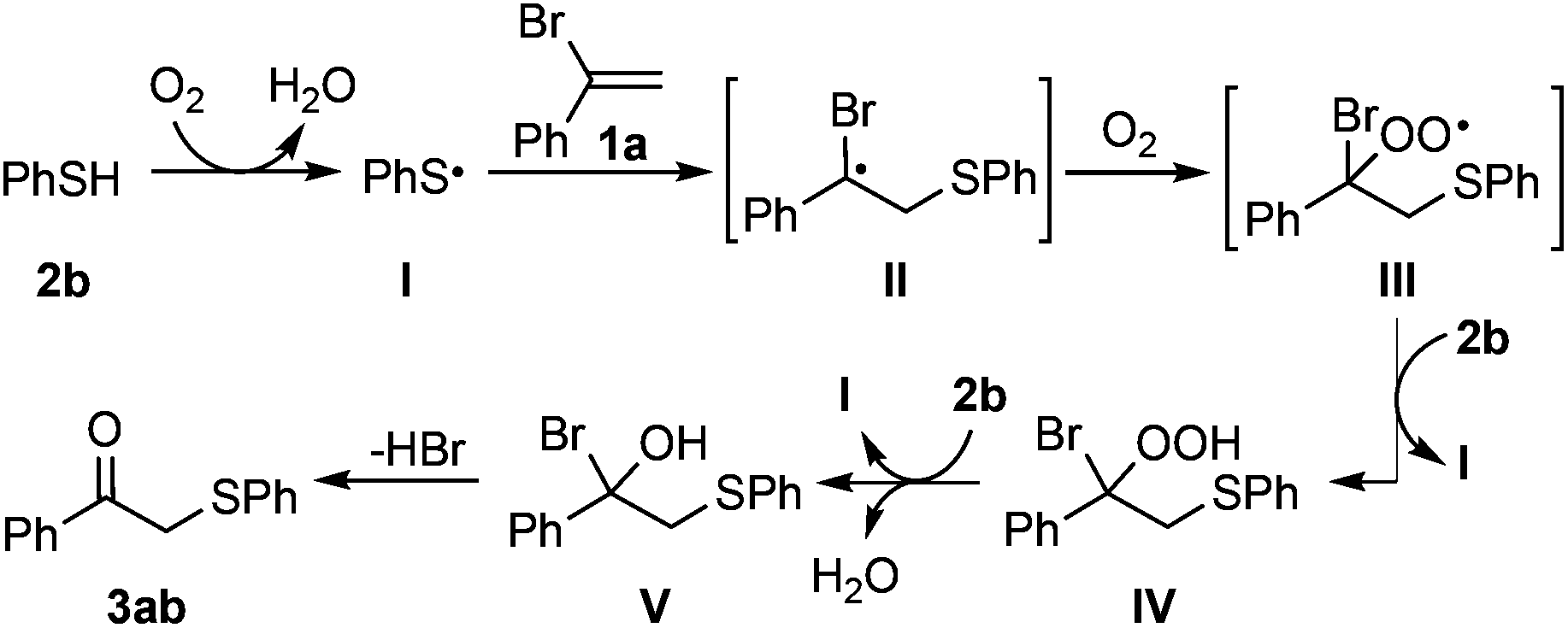 | ||
| Scheme 5 Tentative mechanism. | ||
Conclusions
In conclusion, we have rationally designed a practical protocol for spontaneous O2 fixation at room temperature, in which a series of valuable β-keto sulfides can be synthesized by just mixing the reactants in solvent without the aid of any external additives. The operational simplicity, mild conditions, and high efficiency are the distinct advantages for the usefulness of this reaction. Mechanistic investigation provides insight into the dioxygen and autoinductive effect that can play vital roles for the design of environmentally friendly and sustainable methods for chemical synthesis. The transformation holds significant potential for the applications to more novel organic reactions. Extending this reaction to other synthetic applications is underway.Acknowledgements
This work was supported by the 973 Program (2012CB725302), the National Natural Science Foundation of China (21390400, 21025206, 21272180, and 21302148), and the Research Fund for the Doctoral Program of Higher Education of China (20120141130002) and the Ministry of Science and Technology of China (2012YQ120060). The Program of Introducing Talents of Discipline to Universities of China (111 Program) is also appreciated.Notes and references
- (a) J. Piera and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2008, 47, 3506 CrossRef CAS PubMed; (b) T. Punniyamurthy, S. Velusamy and J. Iqbal, Chem. Rev., 2005, 105, 2329 CrossRef CAS PubMed; (c) Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381 RSC; (d) E. I. Solomon, P. Chen, M. Metz, S.-K. Lee and A. E. Palmer, Angew. Chem., Int. Ed., 2001, 40, 4570 CrossRef CAS; (e) A. E. Wendlandt, A. M. Suess and S. S. Stahl, Angew. Chem., Int. Ed., 2011, 50, 11062 CrossRef CAS PubMed; (f) W. Wu and H. Jiang, Acc. Chem. Res., 2012, 45, 1736 CrossRef CAS PubMed.
- (a) S. Chiba, L. Zhang and J.-Y. Lee, J. Am. Chem. Soc., 2010, 132, 7266 CrossRef CAS PubMed; (b) G. J. Chuang, W. Wang, E. Lee and T. Ritter, J. Am. Chem. Soc., 2011, 133, 1760 CrossRef CAS PubMed; (c) S. O. Kim, C. V. Sastri, M. S. Seo, J. Kim and W. Nam, J. Am. Chem. Soc., 2005, 127, 4178 CrossRef CAS PubMed; (d) C. A. Lippert, S. A. Arnstein, C. D. Sherrill and J. D. Soper, J. Am. Chem. Soc., 2010, 132, 3879 CrossRef CAS PubMed; (e) C. J. Rolle, K. I. Hardcastle and J. D. Soper, Inorg. Chem., 2008, 47, 1892 CrossRef CAS PubMed; (f) Y. Su, X. Sun, G. Wu and N. Jiao, Angew. Chem., Int. Ed., 2013, 52, 9808 CrossRef CAS PubMed; (g) T. Taniguchi, Y. Sugiura, H. Zaimoku and H. Ishibashi, Angew. Chem., Int. Ed., 2010, 49, 10154 CrossRef CAS PubMed; (h) K. K. Toh, Y.-F. Wang, E. P. J. Ng and S. Chiba, J. Am. Chem. Soc., 2011, 133, 13942 CrossRef CAS PubMed; (i) A. Wang and H. Jiang, J. Am. Chem. Soc., 2008, 130, 5030 CrossRef CAS PubMed; (j) H. Wang, Y. Wang, D. Liang, L. Liu, J. Zhang and Q. Zhu, Angew. Chem., Int. Ed., 2011, 50, 5678 CrossRef CAS PubMed; (k) T. Wang and N. Jiao, J. Am. Chem. Soc., 2013, 135, 11692 CrossRef CAS PubMed; (l) Y.-F. Wang, H. Chen, X. Zhu and S. Chiba, J. Am. Chem. Soc., 2012, 134, 11980 CrossRef CAS PubMed; (m) W. Wei and J.-X. Ji, Angew. Chem., Int. Ed., 2011, 50, 9097 CrossRef CAS PubMed; (n) Z. Xu, C. Zhang and N. Jiao, Angew. Chem., Int. Ed., 2012, 51, 11367 CrossRef CAS PubMed; (o) Y. Yan, P. Feng, Q.-Z. Zheng, Y.-F. Liang, J.-F. Lu, Y. Cui and N. Jiao, Angew. Chem., Int. Ed., 2013, 52, 5827 CrossRef CAS PubMed; (p) C. Zhang, P. Feng and N. Jiao, J. Am. Chem. Soc., 2013, 135, 15257 CrossRef CAS PubMed; (q) C. Zhang and N. Jiao, J. Am. Chem. Soc., 2009, 132, 28 CrossRef PubMed; (r) Y.-H. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 14654 CrossRef CAS PubMed; (s) Y.-Q. Zou, J.-R. Chen, X.-P. Liu, L.-Q. Lu, R. L. Davis, K. A. Jørgensen and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 784 CrossRef CAS PubMed.
- C. Ollivier and P. Renaud, Chem. Rev., 2001, 101, 3415 CrossRef CAS PubMed.
- (a) B. C. Giglio, V. A. Schmidt and E. J. Alexanian, J. Am. Chem. Soc., 2011, 133, 13320 CrossRef CAS PubMed; (b) Q. Lu, J. Zhang, F. Wei, Y. Qi, H. Wang, Z. Liu and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 7156 CrossRef CAS PubMed; (c) Y. Nobe, K. Arayama and H. Urabe, J. Am. Chem. Soc., 2005, 127, 18006 CrossRef CAS PubMed; (d) V. A. Schmidt and E. J. Alexanian, Angew. Chem., Int. Ed., 2010, 49, 4491 CrossRef CAS PubMed; (e) V. A. Schmidt and E. J. Alexanian, Chem. Sci., 2012, 3, 1672 RSC; (f) Q. Lu, J. Chen, C. Liu, Z. Huang, P. Peng, H. Wang and A. Lei, RSC Adv., 2015, 5, 24494 RSC.
- Y.-F. Liang and N. Jiao, Angew. Chem., Int. Ed., 2014, 53, 548 CrossRef CAS PubMed.
- (a) V. Chudasama, R. J. Fitzmaurice, J. M. Ahern and S. Caddick, Chem. Commun., 2010, 46, 133 RSC; (b) V. Chudasama, R. J. Fitzmaurice and S. Caddick, Nat. Chem., 2010, 2, 592 CrossRef CAS PubMed.
- H. Togo, Advanced Free Radical Reactions for Organic Synthesis, 2004, 1–56 Search PubMed.
- (a) A. Correa, M. Carril and C. Bolm, Angew. Chem., Int. Ed., 2008, 47, 2880 CrossRef CAS PubMed; (b) F. Dénès, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587 CrossRef PubMed; (c) V. T. D'Souza, R. Nanjundiah, J. Baeza H and H. H. Szmant, J. Org. Chem., 1987, 52, 1720 CrossRef; (d) M. S. Kharasch, W. Nudenberg and G. J. Mantell, J. Org. Chem., 1951, 16, 524 CrossRef CAS; (e) X. B. Li, Z. J. Li, Y. J. Gao, Q. Y. Meng, S. Yu, R. G. Weiss, C. H. Tung and L. Z. Wu, Angew. Chem., Int. Ed., 2014, 53, 2085 CrossRef CAS PubMed; (f) K. C. Majumdar and P. Debnath, Tetrahedron, 2008, 64, 9799 CrossRef CAS PubMed; (g) P. C. Montevecchi, M. L. Navacchia and P. Spagnolo, J. Org. Chem., 1997, 62, 5846 CrossRef CAS; (h) L. Rout, T. K. Sen and T. Punniyamurthy, Angew. Chem., Int. Ed., 2007, 46, 5583 CrossRef CAS PubMed; (i) U. Wille, Chem. Rev., 2012, 113, 813 CrossRef PubMed.
- No desired product was detected when 1-bromo-1-butene or benzyl mercaptan was utilized.
- (a) J. Jin, L. Wu and Z. Zhang, Magn. Reson. Chem., 2002, 40, 346 CrossRef CAS PubMed; (b) H. Zhang, Y. Xu, J. Joseph and B. Kalyanaraman, J. Biol. Chem., 2005, 280, 40684 CrossRef CAS PubMed.
- (a) S.-r. Guo, W.-m. He, J.-n. Xiang and Y.-q. Yuan, Chem. Commun., 2014, 50, 8578 RSC; (b) X. Zhu, Y. Shi, H. Mao, Y. Cheng and C. Zhu, Adv. Synth. Catal., 2013, 355, 3558 CrossRef CAS PubMed.
- No desired product 3aa was observed when diary disulfide (1,2-di-ptolyldisulfane) and 1a were subjected to the standard conditions.
- For examples of TEMPO as a combined radical and oxygen source, see: (a) M. Hartmann, Y. Li and A. Studer, J. Am. Chem. Soc., 2012, 134, 16516 CrossRef CAS PubMed; (b) Y. Li and A. Studer, Angew. Chem., Int. Ed., 2012, 51, 8221 CrossRef CAS PubMed; (c) B. Zhang and A. Studer, Org. Lett., 2013, 15, 4548 CrossRef CAS PubMed.
- The formation of 3ab through an intermolecular nucleophilic substitution by H2O cannot be excluded.
- On submission of this work, a similar work on the TBHP triggered radical hydroxysulfurization of styrenes was reported: S.-F. Zhou, X. Pan, Z.-H. Zhou, A. Shoberu and J.-P. Zou, J. Org. Chem, 2015, 80, 3682 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00102a |
| This journal is © the Partner Organisations 2015 |