
Sulfur-containing stable five-membered “cycloallene” complexes: 1-thia-2-zircona- and 1-thia-2-titanacyclopenta-3,4-dienes†‡
Noriyuki
Suzuki
*,
Takumi
Asada
,
Akiko
Kawamura
and
Yoshiro
Masuyama
Department of Materials and Life Sciences, Faculty of Science and Technology, Sophia University, 7-1 Kioi-cho, Chiyoda-ku, Tokyo 102-8554, Japan. E-mail: norisuzuki@sophia.ac.jp
First published on 13th April 2015
Abstract
Stable five-membered metallacyclic allenes that contain a sulfur atom were synthesized and structurally characterized. Low-valent zirconocene and titanocene reacted with alkynylthioamides to afford 1-thia-2-metallacyclopenta-3,4-diene compounds in moderate to excellent yields. The molecular structure of the zirconium complexes indicated their cycloallene structure, while an η2-alkyne-coordinated structure might be more responsible for the titanium complexes. Hydrolysis of the compounds gave the corresponding (Z)-alkenylthioamides predominantly. Deuterolysis resulted in >99% D incorporation.
Introduction
Allene compounds have attracted organic chemists because they exhibit a variety of reactivities and are versatile for organic synthesis.1 Cyclic allenes are more reactive than linear allenes, and are usually unstable, especially for small ring sizes. Cyclopenta-1,2-dienes, which are five-membered cyclic allenes, have been reported to exist as very short-lived intermediates.2 Cycloallenes can be stabilized by replacing one or more of the ring members with heteroatoms, such as silicon or phosphorus. A few six-membered heterocyclic allenes have been isolated, and their molecular structures have been unambiguously determined.3Recently, our group and Erker's group have independently reported the synthesis of metal-containing five-membered cycloallenes (1 and 2, Fig. 1). It was surprising that these compounds were very stable despite their highly strained allene structures, and the molecular structures were unambiguously determined.4 Rosenthal and coworkers demonstrated the synthesis of nitrogen-containing five-membered metallacycloallene compounds, 1-aza-2-metallacyclopenta-3,4-dienes 3, by C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N insertion into the Zr–C bond, followed by silyl group-rearrangement.5
N insertion into the Zr–C bond, followed by silyl group-rearrangement.5
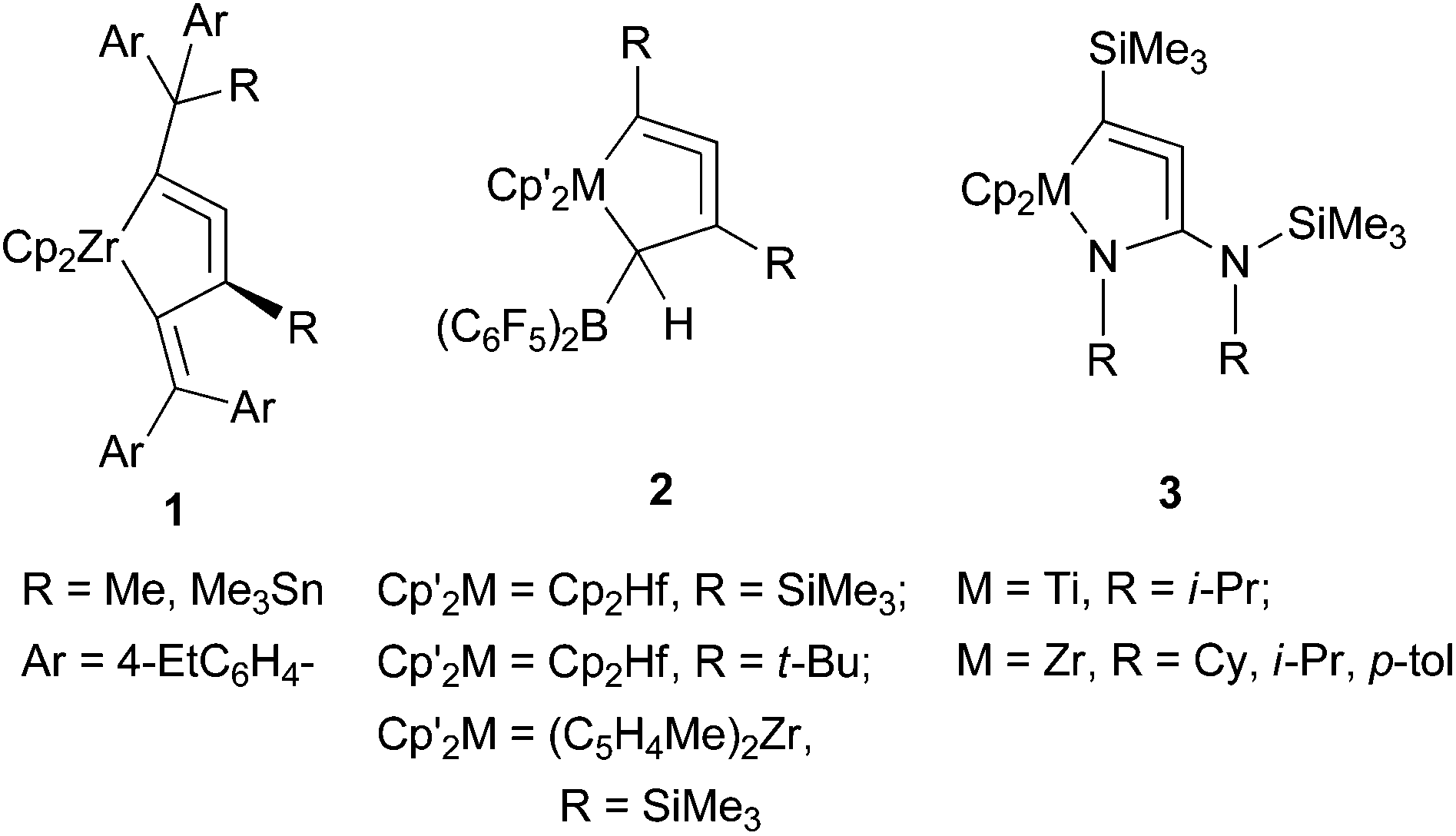 | ||
| Fig. 1 Stable five-membered metallacycloallenes. | ||
However, the preparative methods of these complexes are not generally applicable for the synthesis of a variety of their analogues. We have reported a simpler protocol to prepare 1-metallacyclopenta-2,3-diene complexes from the reaction of a low-valent zirconocene with 1-en-3-yne compounds.6 Erker and coworkers have described an alternative approach by borane-catalyzed reductive elimination from (σ-alkynyl)(σ-alkenyl)zirconocene complexes.7 Alternative preparative methods for 1-aza-2-zirconacyclopenta-3,4-diene have also been reported.8
We have demonstrated in these previous reports that 1-zirconacyclopenta-2,3-dienes and 1-aza-2-zirconacyclopenta-3,4-dienes can be prepared from the reaction of low-valent zirconocenes with 1-en-3-ynes and alkynylimines, respectively (Scheme 1). This protocol seems to be applicable to other types of metallacycloallenes. To the best of our knowledge, 1-aza-2-zirconacyclopenta-3,4-dienes are the only examples for reported stable five-membered metallacycloallenes containing a heteroatom. These results prompted us to explore the possibility of synthesizing sulfur-containing analogues of these compounds as well as other group 4 metal complexes. We herein report the synthesis and structure of sulfur-containing zirconacycloallenes and titanacycloallenes.
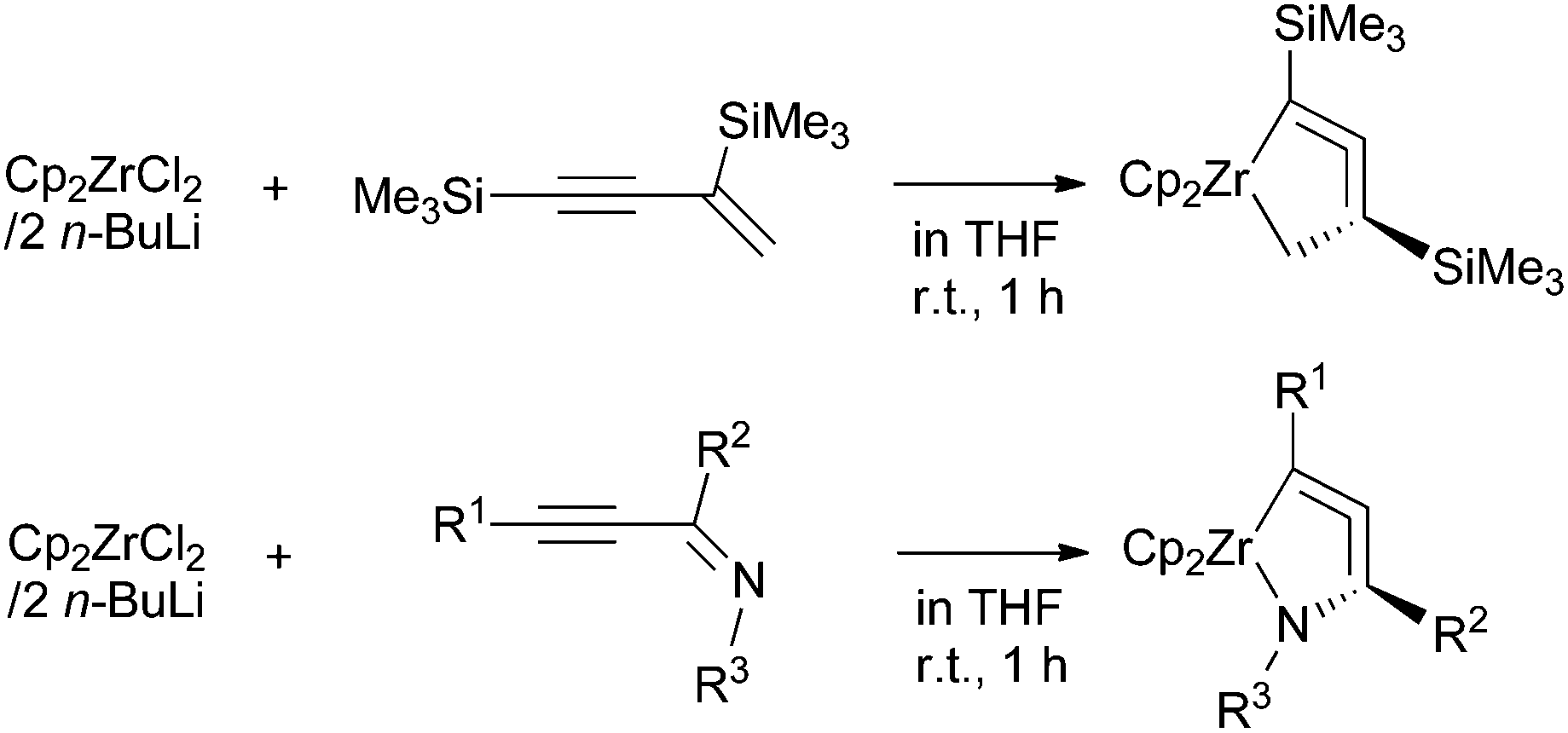 | ||
| Scheme 1 Simple synthetic protocols for metallacycloallenes. | ||
Results and discussion
In our previous study on 1-aza-2-zirconacyclopenta-3,4-dienes, we employed alkynylimines as the starting materials. To synthesize the sulfur-analogues, we chose alkynylthioamides as starting compounds because these are rather stable sulfur-derivatives of 1-en-3-ynes.9We added N,N-dimethyl-3-(trimethylsilyl)prop-2-ynethioamide (4a) to di-n-butylzirconocene, Negishi reagent,10 that was generated in situ from zirconocene dichloride and two equiv. of n-butyllithium at −78 °C. After stirring at room temperature for 1 h, the formation of 1-thia-2-zirconacyclopenta-3,4-diene 5a was observed by 1H NMR spectroscopy (69% yield) (Scheme 2). Two singlets assigned to cyclopentadienyl (Cp) ligands appeared at 5.25 and 5.49 ppm. The methyl groups on the nitrogen atom were observed as a single signal at 2.59 ppm, while the two methyl groups were observed to be inequivalent for 4a. N,N-Diethyl-3-(tert-butyldimethylsilyl)prop-2-ynethioamide (4b) gave 5b in 83% yield. The molecular structures of 5a and 5b were determined by X-ray diffraction analyses, although the quality of the structural data of 5a was unsatisfactory to discuss its bond lengths and angles in detail.11 These are the first examples of stable five-membered metallacycloallenes that contain a sulfur atom. These complexes are considerably stable at room temperature under an inert atmosphere. For example, crystals of 5b can be kept at room temperature for a month under argon. Trialkylsilyl groups were found to be suitable as R substituents. We tried to synthesize alkynyl thioamides with R = phenyl group, but the reactions gave a complex mixture.
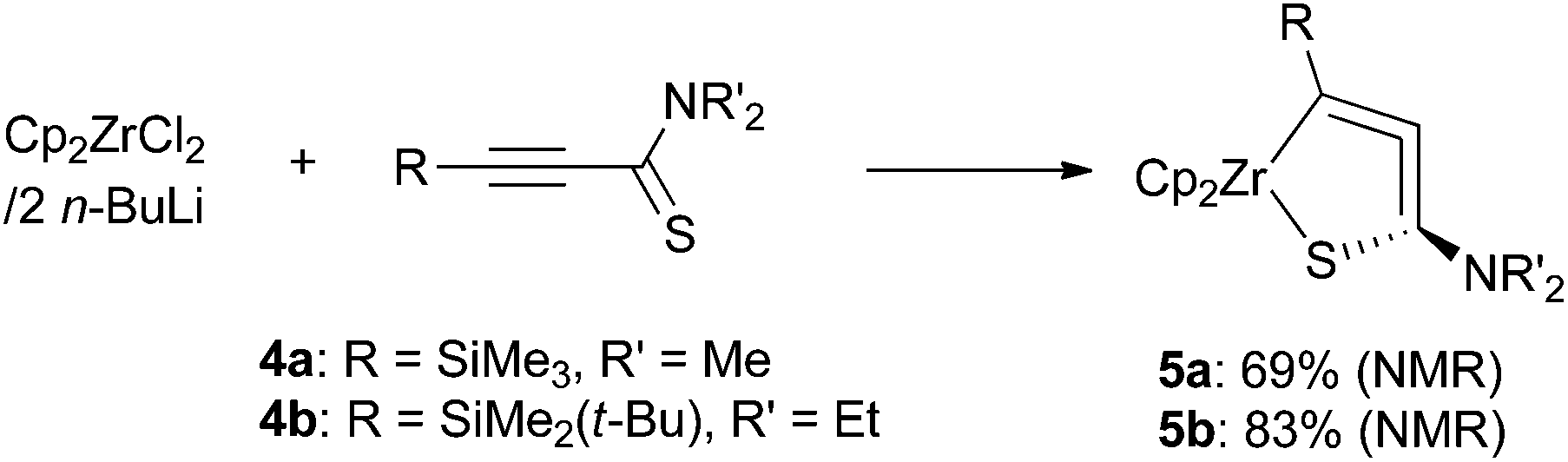 | ||
| Scheme 2 Synthesis of 1-thia-2-zirconacyclopenta-3,4-dienes. | ||
Starting from Cp2TiCl2 and two equiv. of n-butyllithium serving as a precursor of “Cp2Ti(II)” species,12 the titanium analogues 6a and 6b were also synthesized in moderate yields (Scheme 3). The yield was improved when two equiv. of titanium was used in the reaction (6b, 58%). Titanium complexes were rather unstable compared with their zirconium analogues. For example, they degraded within a day at room temperature in a dry C6D6 solution, even under an argon atmosphere.
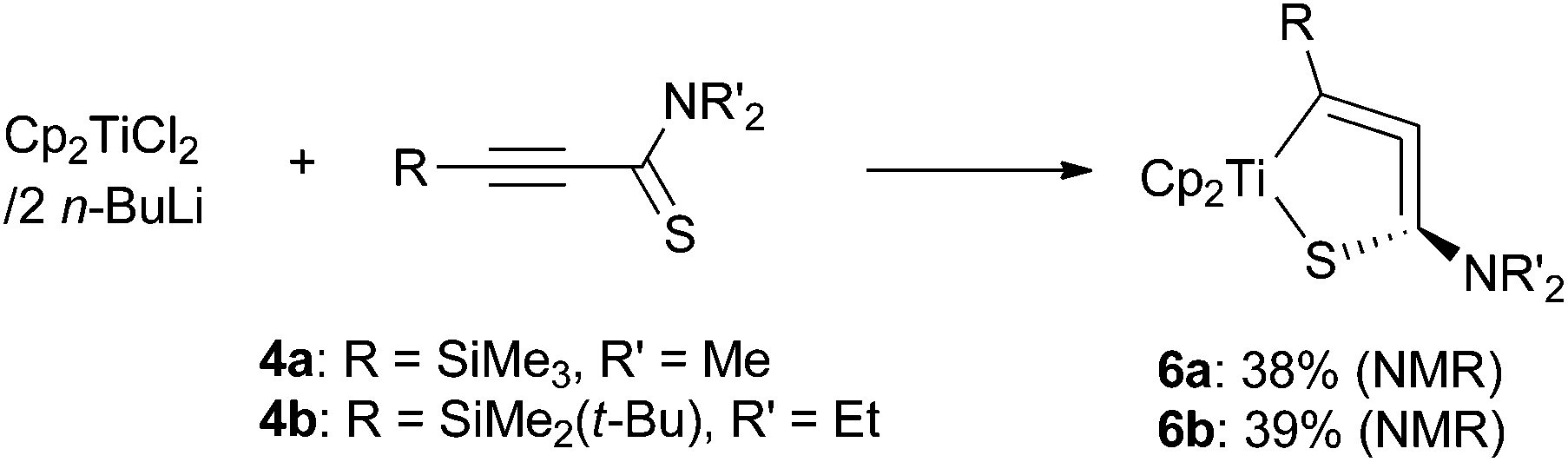 | ||
| Scheme 3 Preparation of 1-thia-2-titanacyclopenta-3,4-dienes. | ||
The zirconium complex 5b and the titanium complex 6b showed similar molecular structures (Fig. 2). It is noteworthy that these two have almost the same cell parameters, indicating that they are nearly isostructural. Selected bond lengths and angles are summarized in Table 1.
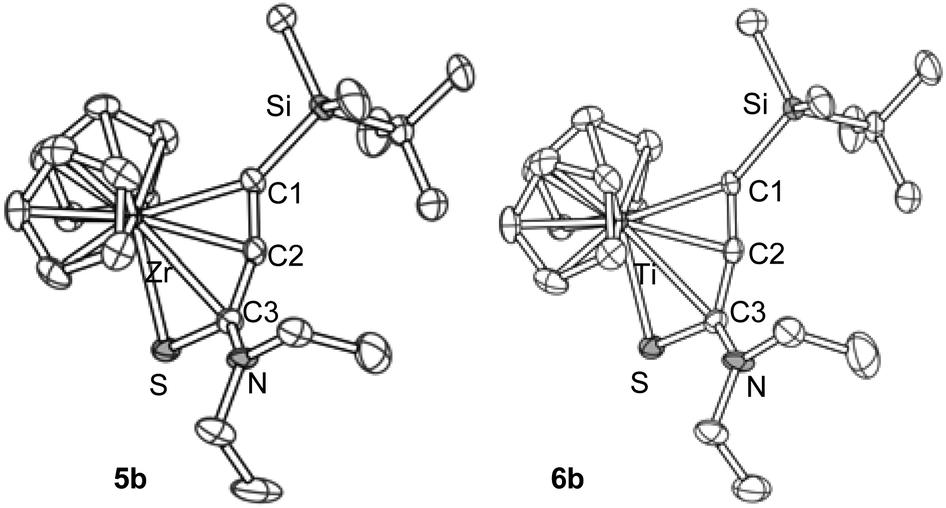 | ||
| Fig. 2 The molecular structures of 5b and 6b. Hydrogen atoms are omitted for clarity. One position of the disordered groups in 5b is omitted. | ||
| 5b (M = Zr) | 6b (M = Ti) | |
|---|---|---|
| M–S | 2.5875(12) | 2.5348(17) |
| M–C1 | 2.306(2) | 2.245(4) |
| M–C2 | 2.342(2) | 2.254(4) |
| M–C3 | 2.655(3) | 2.671(5) |
| C1–C2 | 1.286(3) | 1.271(6) |
| C2–C3 | 1.377(3) | 1.381(5) |
| S–C3 | 1.745(2) | 1.718(4) |
| Si–C1 | 1.860(2) | 1.850(4) |
| N–C3 | 1.394(3) | 1.375(5) |
| C1–C2–C3 | 158.8(2) | 164.1(4) |
| S–C3–C2 | 115.9(2) | 113.3(3) |
| M–C1–C2 | 75.53(12) | 74.0(2) |
| M–S–C3 | 72.65(7) | 75.08(16) |
| S–M–C1 | 94.87(8) | 96.22(12) |
| S–C3–N | 122.8(2) | 124.5(3) |
| N–C3–C2 | 119.7(2) | 121.2(3) |
The C1–C2 bond distances were 1.27–1.29 Å, showing that these are in the range of short double bonds.13 The C2–C3 bond lengths were 1.37–1.38 Å. The metallacycle structures are not planar and are bent at the M–C3 bonds. These showed similar structural features to the aza-analogues.5a,8 The angle C1–C2–C3 was 159° for the zirconium complex 5b, and was 164° for the titanium complex 6b, showing that the allene moieties are bent from 180°. Note that the M–C3 distance was significantly longer than that reported for 1-zirconacyclopenta-2,3-dienes and 1-aza-2-metallacyclopenta-3,4-dienes.4a,5,7 Two possible structures, P and Q, may contribute to 5 and 6, the 1-thia-2-metallacyclopenta-3,4-diene structure, P, and the η2-alkyne-κS coordination mode, Q (Fig. 3). The S–C3 bond lengths were more than 1.7 Å, and these are closer to the Csp2–S single bonds than the CS double bonds.13
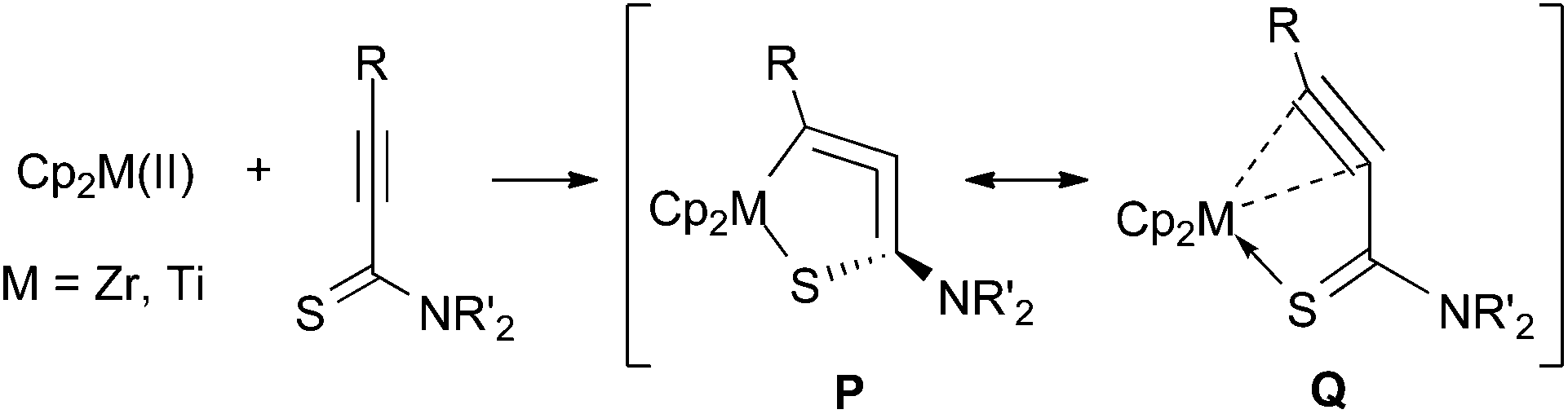 | ||
| Fig. 3 Resonance structures of metallacycloallenes. | ||
The Zr–C1 (2.308 Å) and Zr–C2 (2.344 Å) bond distances in 5b were significantly longer than those reported for Zr-η2-alkyne complexes, where the Zr–C bond distances are 2.18–2.26 Å.14 The reported Zr–S covalent bond lengths in zirconium thiolates are 2.5–2.6 Å, while Zr←S dative bonds are typically 2.7–2.8 Å.15 Thus, the Zr–S bond in 5b (2.588 Å) can be regarded as a covalent bond. These observations suggest that the zirconium compounds 5 should be described with the structure P.
On the other hand, the Ti–C1 and Ti–C2 distances (2.245 and 2.254 Å, respectively) in 6b were not much longer than those reported for Ti-η2-alkyne complexes, where the Ti–C distances are 2.10–2.27 Å.16 The C1–C2 bond length in 6b was slightly shorter than that in the zirconium complex 5b. The Ti–S bond length in 6b was 2.535 Å, which is significantly longer than the reported Ti–S bond lengths in titanium thiolates, where the typically observed values are 2.3–2.4 Å.17 These results are consistent with the fact that the Ti–C1 and Ti–S bond lengths are shorter than the Zr–C1 and Zr–S bonds by only 0.05–0.06 Å, although titanium has a smaller atomic radius than zirconium by more than 0.1 Å (Ti: 1.324 Å, Zr: 1.454 Å).18 These facts suggest that the contribution of Q is more responsible in titanium complexes 6. The shorter S–C3 bond corresponds to this idea.
The chemical shifts in 13C NMR spectroscopy for 5 and 6 are shown in Table 2 along with other metallacycloallenes 7–10 for comparison. A coupling between C1 and the silicon atom was observed (for example, 1JCSi = 67 Hz for 5a). To assign the other quaternary carbon atoms C2 and C3, two dimensional INADEQUATE measurements were carried out on 5a. The central carbon atom of “allene” (C2) appeared at 136.7 ppm, and the coupling constants for C1–C2 and C2–C3 were 1JCC = 68.2 and 88.5 Hz, respectively. These values are similar to those observed in our previous study on zirconacycloallene,4a but are smaller than typical values of acetylene (170 Hz), and are in between the range of ethylene (68 Hz) and allenes (99–103 Hz).19 The chemical shifts for the “allene” moieties in 5 and 6 were observed in the same range as those of the known metallacycloallenes.5a,6a,8 For example, the signals for C2 atoms appeared at 136–138 ppm, although the central carbon atoms of most linear allenes are observed at around 200 ppm.
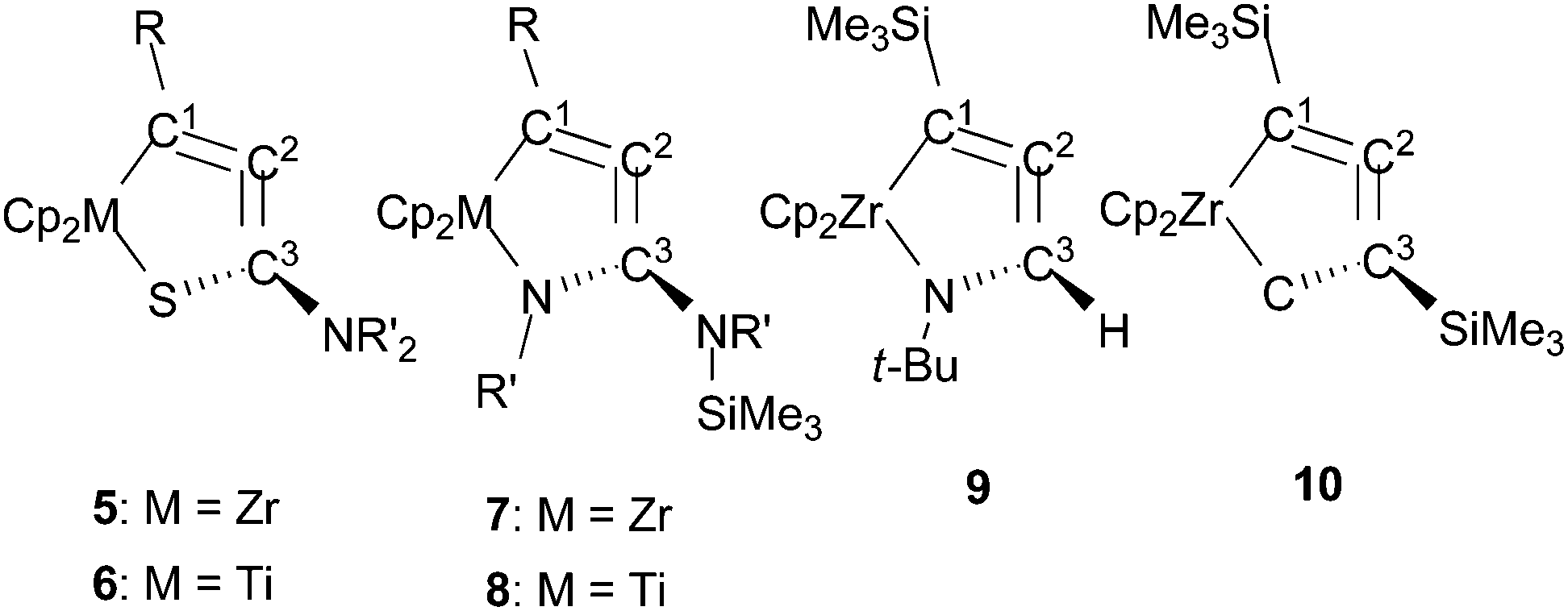
A contribution of the structure Q to the titanium complexes 6 was suggested based on our X-ray diffraction analyses. Nevertheless, the 13C NMR chemical shifts of C1 and C2 in 6 were in the range 136–143 ppm, and were observed at higher field compared with η2-coordinated trimethylsilyl alkyne groups in titanium complexes (196–220 ppm),16a,20 indicating that contribution from the structure P cannot be ruled out.
In the titanium complexes 6, the Cp groups were observed as broad signals at room temperature, as well as methyl/methylene protons on the nitrogen atoms. The Cp signals appeared as two sharp singlets at −40 °C. These coalesced at 40 °C and then sharpen at 60 °C. For the zirconium complex 5b, however, the Cp ligands appeared as two sharp singlets at room temperature, and these did not broaden even at 100 °C. This indicates that the titanacyclic structures flip faster than the corresponding zirconacycles. The two Cp rings are inequivalent because of the enantiotopic structure of the allene, and a fast epimerization leads to broadening of their 1H NMR signals (Fig. 4). The ring flipping must formally involve a bond cleavage of the ring structure, and it is proposed that the η2-alkyne-κS-structure (Q) has a higher contribution in the titanium complexes.
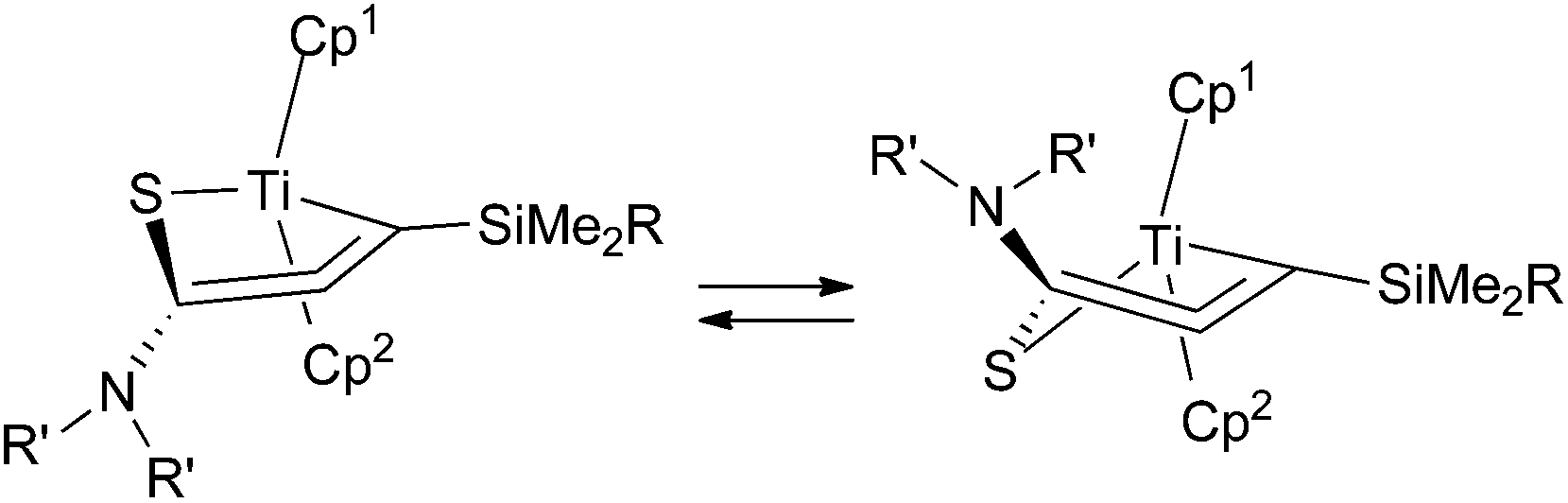 | ||
| Fig. 4 “Ring flipping” in the titanium complex 6. | ||
Erker and coworkers have reported on hafnium and zirconium cycloallene complexes, where diastereoisomerization by ring flipping was faster for the zirconacycle than for the hafnacycle.4b,c
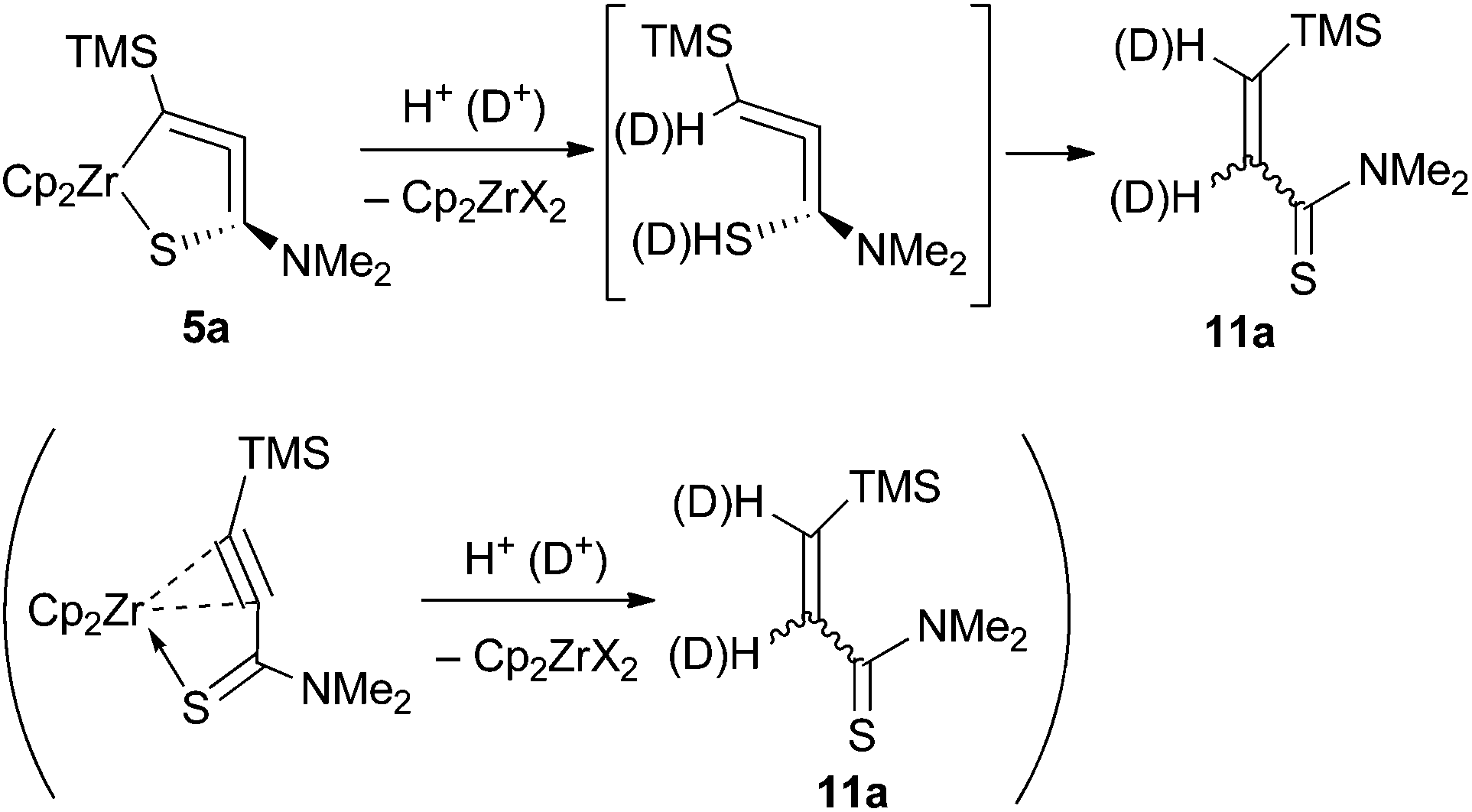
Hydrolysis of 5a with dil. HCl gave the corresponding Z-alkene 11a in an 86![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :14 Z/E ratio (76% combined yield, Scheme 4). It has been reported that hydrolysis of zirconium-alkyne complexes affords the corresponding Z-alkenes.10a,21 The deuterolysis of 5a using a diethyl ether solution of DCl gave the dideuterated alkene with >99% incorporation of deuterium. Complex 5b also gave 11b after hydrolysis.
:14 Z/E ratio (76% combined yield, Scheme 4). It has been reported that hydrolysis of zirconium-alkyne complexes affords the corresponding Z-alkenes.10a,21 The deuterolysis of 5a using a diethyl ether solution of DCl gave the dideuterated alkene with >99% incorporation of deuterium. Complex 5b also gave 11b after hydrolysis.
 | ||
| Scheme 4 Hydrolysis of 5a. | ||
Hydrolysis might have occurred at the 1,4-position of the metallacycles to give a formal “allenylaminothiol” followed by tautomerization into 11. Another possibility is that hydrolysis took place at the 3,4-position, i.e., the alkyne carbons, to afford the alkene. Part of the Z-alkene might have isomerized into the trans-alkene.22 Further investigation on the synthesis of the analogues, and reactivity of the complexes is now in progress.
Experimental
General
All manipulations involving organometallic compounds were conducted under an inert atmosphere using standard Schlenk techniques or a glove box. The anhydrous tetrahydrofuran was purchased from Kanto Chemical Co., Inc. and purified using a Glass Contour Solvent System™ (SG Water USA). Hexane and toluene (dehydrated grade) were purchased from Kanto Chemical Co., Inc., degassed and kept under argon. n-Butyllithium (hexane solution) were purchased from Kanto Chemical Co., Inc. Dichlorobis(η5-cyclopentadienyl)zirconium and N,N-diethylthiocarbamoyl chloride were purchased from Sigma-Aldrich Corp. Trimethylsilylacetylene was purchased from Tokyo Chemical Industry Co., Ltd. N,N-Dimethylthiocarbamoyl chloride and tert-butyldimethylsilylacetylene were obtained from Wako Pure Chemical Industries Ltd. These chemicals were used as received unless otherwise mentioned. N,N-Dimethyl-3-(trimethylsilyl)prop-2-ynethioamide (4a) was prepared according to a literature method.9a The INADEQUATE measurement was recorded on a JNM-ECA 600 spectrometer equipped with an UltraCOOL probe (JEOL RESONANCE). The other NMR spectra were recorded on JNM-ECX 300 and ECA 500 spectrometers (JEOL), and NMR yields were determined using anhydrous toluene as an internal standard. Infrared spectra were recorded on a Shimadzu FT-IR 8300. EI-MS spectra were recorded on a Shimadzu GCMS QP-5050 instrument or on a JEOL JMS-700 instrument. ESI-MS spectra were recorded on a JEOL JMS-T100LC instrument or on a Thermo Exactive spectrometer. Elemental analyses were performed on a Perkin Elmer PE2400 Series II CHNS/O.![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C), 102.08 (CC), 176.73 (CS). IR (KBr): 2858, 2932, 2149, 1655, 1628, 1489, 1423, 1277, 1123, 1007, 779, 671 cm−1. Anal. calcd for C13H25NSSi, C 61.11, H 9.86, N 5.48, S 12.55; found C 60.77, H 9.79, N 5.58, S 12.39.
CC), 140.70 (q, 1JCC = 68.2 Hz, 1JCSi = 67 Hz, CSiMe3). IR (KBr): 664, 849, 1092, 1261, 1801 cm−1. High resolution mass spectrometry: calcd for C18H25NSSiZr + H+ [M + H]+, 406.0602, found, 406.0603.
CC). IR (KBr): 799, 1015, 1088, 1231, 1493, 1628, 1674, 1805, 3086, 3807 cm−1. Elemental analysis, calcd for C23H35NSSiZr, C 57.92, H 7.40, N 2.94, S 6.72; found C 57.93, H 7.41, N 2.91, S 6.72.
CC), 142.52 (q, 1JCSi = 73 Hz, CSi).
CC), 138.05 (q, 1JCSi = 70 Hz, CSi).
C), 102.08 (CC), 176.73 (CS). IR (KBr): 2858, 2932, 2149, 1655, 1628, 1489, 1423, 1277, 1123, 1007, 779, 671 cm−1. Anal. calcd for C13H25NSSi, C 61.11, H 9.86, N 5.48, S 12.55; found C 60.77, H 9.79, N 5.58, S 12.39.
CC), 140.70 (q, 1JCC = 68.2 Hz, 1JCSi = 67 Hz, CSiMe3). IR (KBr): 664, 849, 1092, 1261, 1801 cm−1. High resolution mass spectrometry: calcd for C18H25NSSiZr + H+ [M + H]+, 406.0602, found, 406.0603.
CC). IR (KBr): 799, 1015, 1088, 1231, 1493, 1628, 1674, 1805, 3086, 3807 cm−1. Elemental analysis, calcd for C23H35NSSiZr, C 57.92, H 7.40, N 2.94, S 6.72; found C 57.93, H 7.41, N 2.91, S 6.72.
CC), 142.52 (q, 1JCSi = 73 Hz, CSi).
CC), 138.05 (q, 1JCSi = 70 Hz, CSi).
(Z)-11a (major). 1H NMR (CDCl3, Me4Si, 500 MHz): δ 0.12 (s, 9H), 3.31 (s, 3H), 3.44 (s, 3H), 5.73 (d, J = 16.0 Hz, 1H), 6.93 (d, J = 16.0 Hz, 1H). 13C NMR (CDCl3, Me4Si, 125.8 MHz): δ −0.67 (CH3Si), 41.89 (CH3N), 42.74 (CH3N), 131.94 (CHSi), 145.26 (CH–CS), 199.48 (q, CS). (E)-11a (minor): 1H NMR (CDCl3, Me4Si, 500 MHz): δ 0.14 (s, 9H), 3.35 (s, 3H), 3.52 (s, 3H), 6.80 (d, J = 18.3 Hz, 1H), 6.96 (d, J = 18.3 Hz, 1H). 13C NMR (500 MHz, CDCl3, Me4Si): δ −1.54 (CH3Si), 42.10 (CH3N), 44.22 (CH3N), 140.55 (CHSi), 144.15 (CH–CS), 197.45 (CS). IR (NaCl, E/Z mixture): 732.9, 983.6, 1199.6, 1350.1, 1454.2, 1593.1, 1643.2, 2954.7. High resolution mass spectrometry (E/Z mixture): [M + H]+m/z calcd for C8H18NSSi 188.0929, found 188.0924.
(Z)-11b (major). 1H NMR (CDCl3, Me4Si, 500 MHz): δ 0.12 (s, 6H), 0.90 (s, 9H), 1.22 (t, J = 6.9 Hz, 3H), 1.30 (t, J = 6.9 Hz, 3H), 3.66 (q, J = 6.9 Hz, 2H), 3.97 (q, J = 6.9 Hz, 2H), 5.75 (d, J = 15.5 Hz, 1H), 7.00 (d, J = 15.5 Hz, 1H). 13C NMR (CDCl3, Me4Si, 125.8 MHz): δ −5.05 (CH3Si), 11.18 (CH3CH2), 12.98 (CH3CH2), 16.66 (q, CSi), 26.50 ((CH3)3C), 45.68 (CH2N), 46.88 (CH2N), 128.82 (CHSi), 146.07 (CHCS), 198.48 (CS). (E)-11b (minor). 1H NMR (CDCl3, Me4Si, 500 MHz): δ 0.10 (s, 6H), 0.91 (s, 9H), 1.27 (t, J = 6.9 Hz, 3H), 1.30(t, J = 6.9 Hz, 3H), 3.65 (q, J = 6.9 Hz, 2H), 4.03 (q, J = 6.9 Hz, 2H), 6.83 (d, J = 18 Hz, 1H), 7.01 (d, J = 18 Hz, 1H). 13C NMR (CDCl3, Me4Si, 125.8 MHz): δ −6.27 (CH3Si), 11.35 (CH3CH2), 13.61 (CH3CH2), 16.77 (q, CSi), 26.45 ((CH3)3C), 46.54 (CH2N), 48.12 (CH2N), 141.47 (CHSi), 141.84 (CHCS), 196.14 (CS). High resolution mass spectrometry (E/Z mixture): [M + H]+m/z calcd for C13H28NSSi 258.1712, found, 258.1741.
Conclusions
Low-valent zirconocene and titanocene reacted with alkynylthioamides to give “1-thia-2-metallacyclopenta-3,4-diene” compounds. X-ray diffraction analyses showed that the zirconium complexes have a sulfur-containing cycloallene structure, while an η2-alkyne-coordinated structure might have more contribution to the titanium complexes. Hydrolysis of the compounds gave the corresponding (Z)-alkenylthioamides as major products.Acknowledgements
This study was financially supported by JSPS Kakenhi (B)22350022 and (C)26410058. We thank Mr Keiichi Yoshida (JEOL RESONANCE) for assistance in INADEQUATE measurement. Dr Daisuke Hashizume (RIKEN) is acknowledged for assistance in X-ray diffraction analysis. We express our gratitude to Ms. Saeko Machishima for assistance in elemental analysis. Efforts in mass spectrometry by Ms Emiko Okano and Dr Jiro Fujishige is greatly appreciated. Prof. Hideki Abe (RIKEN) is acknowledged for his valuable advice.Notes and references
- (a) M. Christl, in Modern Allene Chemistry, ed. N. Krause and A. S. K. Hashmi, Wiley-VCH, Weinheim, 2004, pp. 243–357 Search PubMed; (b) M. Ogasawara, in Cumulenes and Allenes, Science of Synthesis, ed. N. Krause, Thieme, Stuttgart, 2007, pp. 9–70 Search PubMed.
- (a) F. Algi, R. Özen and M. Balci, Tetrahedron Lett., 2002, 43, 3129–3131 CrossRef CAS; (b) M. Ceylan, S. Yalçin, H. Seçen, Y. Sütbeyaz and M. Balci, J. Chem. Res., 2003, 21–23 CrossRef.
- (a) Y. Pang, S. A. Petrich, V. G. Young Jr., M. S. Gordon and T. J. Barton, J. Am. Chem. Soc., 1993, 115, 2534–2536 CrossRef CAS; (b) T. Shimizu, F. Hojo and W. Ando, J. Am. Chem. Soc., 1993, 115, 3111–3115 CrossRef CAS; (c) M. A. Hofmann, U. Bergsträsser, G. J. Reiss, L. Nyulászi and M. Regitz, Angew. Chem., Int. Ed., 2000, 39, 1261–1263 CrossRef CAS.
- (a) N. Suzuki, D. Hashizume, H. Koshino and T. Chihara, Angew. Chem., Int. Ed., 2008, 47, 5198–5202 CrossRef CAS PubMed; (b) J. Ugolotti, G. Dierker, G. Kehr, R. Fröhlich, S. Grimme and G. Erker, Angew. Chem., Int. Ed., 2008, 47, 2622–2625 CrossRef CAS PubMed; (c) J. Ugolotti, G. Kehr, R. Fröhlich, S. Grimme and G. Erker, J. Am. Chem. Soc., 2009, 132, 1996–2007 CrossRef PubMed; (d) N. Suzuki and D. Hashizume, Coord. Chem. Rev., 2010, 254, 1307–1326 CrossRef CAS PubMed.
- (a) K. Kaleta, M. Ruhmann, O. Theilmann, T. Beweries, S. Roy, P. Arndt, A. Villinger, E. D. Jemmis, A. Schulz and U. Rosenthal, J. Am. Chem. Soc., 2011, 133, 5463–5473 CrossRef CAS PubMed; (b) S. Roy, E. D. Jemmis, M. Ruhmann, A. Schulz, K. Kaleta, T. Beweries and U. Rosenthal, Organometallics, 2011, 30, 2670–2679 CrossRef CAS.
- (a) N. Suzuki, T. Shimura, Y. Sakaguchi and Y. Masuyama, Pure Appl. Chem., 2011, 83, 1781–1788 CrossRef CAS. See also: (b) G. Bender, G. Kehr, R. Fröhlich, J. L. Petersen and G. Erker, Chem. Sci., 2012, 3, 3534–3540 RSC.
- S. K. Podiyanachari, G. Bender, C. G. Daniliuc, G. Kehr and G. Erker, Organometallics, 2014, 33, 3481–3488 CAS.
- S. K. Podiyanachari, R. Fröhlich, C. G. Daniliuc, J. L. Petersen, G. Kehr, G. Erker, N. Suzuki, S. Yuasa, K. Hagimori, S. Inoue, T. Asada, T. Takemoto and Y. Masuyama, Dalton Trans., 2012, 41, 10811–10816 RSC.
- (a) K. Hartke, H.-D. Gerber and U. Roesrath, Liebigs Ann. Chem., 1991, 903–916 CrossRef CAS PubMed; (b) K. Hartke, H.-D. Gerber and U. Roesrath, Tetrahedron Lett., 1989, 30, 1073–1076 CrossRef CAS.
- (a) E.-i. Negishi, S. J. Holmes, J. M. Tour, J. A. Miller, F. E. Cederbaum, D. R. Swanson and T. Takahashi, J. Am. Chem. Soc., 1989, 111, 3336–3346 CrossRef CAS; (b) E.-i. Negishi, F. E. Cederbaum and T. Takahashi, Tetrahedron Lett., 1986, 27, 2829–2832 CrossRef CAS.
- See the ESI.‡.
- K. Sato, Y. Nishihara, S. Huo, Z. Xi and T. Takahashi, J. Organomet. Chem., 2001, 633, 18–26 CrossRef CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- (a) U. Rosenthal, A. Ohff, M. Michalik, H. Görls, V. V. Burlakov and V. B. Shur, Angew. Chem., Int. Ed. Engl., 1993, 32, 1193–1195 CrossRef PubMed; (b) A. K. List, K. Koo, A. L. Rheingold and G. L. Hillhouse, Inorg. Chim. Acta, 1998, 270, 399–404 CrossRef CAS.
- (a) S. L. Buchwald, R. B. Nielsen and J. C. Dewan, J. Am. Chem. Soc., 1987, 109, 1590–1591 CrossRef CAS; (b) M. J. Carney, P. J. Walsh and R. G. Bergman, J. Am. Chem. Soc., 1990, 112, 6426–6428 CrossRef CAS; (c) J. J. W. Eshuis, Y. Y. Tan, A. Meetsma, J. H. Teuben, J. Renkema and G. G. Evens, Organometallics, 1992, 11, 362–369 CrossRef CAS; (d) R. Fandos, M. Lanfranchi, A. Otero, M. A. Pellinghelli, M. J. Ruiz and P. Terreros, Organometallics, 1996, 15, 4725–4730 CrossRef CAS; (e) P. Arndt, C. Lefeber, R. Kempe and U. Rosenthal, Chem. Ber., 1996, 129, 207–211 CrossRef CAS PubMed; (f) E. Ortega, N. Pirio, P. Meunier and B. Donnadieu, Chem. Commun., 2004, 678–679 RSC; (g) S. Bredeau, E. Ortega, G. Delmas, P. Richard, R. Fröhlich, B. Donnadieu, G. Kehr, N. Pirio, G. Erker and P. Meunier, Organometallics, 2009, 28, 181–187 CrossRef CAS.
- (a) A. Ohff, P. Kosse, W. Baumann, A. Tillack, R. Kempe, H. Goerls, V. V. Burlakov and U. Rosenthal, J. Am. Chem. Soc., 1995, 117, 10399–10400 CrossRef CAS; (b) P. Štěpnička, R. Gyepes, I. Císařová, V. Varga, M. Polášek, M. Horáček and K. Mach, Organometallics, 1999, 18, 627–633 CrossRef.
- (a) P. R. Stafford, T. B. Rauchfuss, A. K. Verma and S. R. Wilson, J. Organomet. Chem., 1996, 526, 203–214 CrossRef CAS; (b) R. Beckhaus, J. Sang, T. Wagner and U. Böhme, J. Chem. Soc., Dalton Trans., 1997, 2249–2256 RSC.
- L. Pauling, J. Am. Chem. Soc., 1947, 69, 542–553 CrossRef CAS.
- (a) J. Kaski, P. Lantto, J. Vaara and J. Jokisaari, J. Am. Chem. Soc., 1998, 120, 3993–4005 CrossRef CAS; (b) O. B. Lutnæs, T. A. Ruden and T. Helgaker, Magn. Reson. Chem., 2004, 42, S117–S127 CrossRef PubMed; (c) A. Rajca and L. M. Tolbert, J. Am. Chem. Soc., 1987, 109, 1782–1789 CrossRef CAS.
- U. Rosenthal, H. Görls, V. V. Burlakov, V. B. Shur and M. E. Vol'pin, J. Organomet. Chem., 1992, 426, C53–C57 CrossRef CAS.
- G. A. Vaughan, G. L. Hillhouse, R. T. Lum, S. L. Buchwald and A. L. Rheingold, J. Am. Chem. Soc., 1988, 110, 7215–7217 CrossRef CAS.
- Zirconocene-catalyzed Z/E isomerization of alkene has been reported, see: E.-i. Negishi, D. Choueiry, T. B. Nguyen, D. R. Swanson, N. Suzuki and T. Takahashi, J. Am. Chem. Soc., 1994, 116, 9751–9752 CrossRef CAS.
- M. C. Burla, M. Camalli, B. Carrozzini, G. L. Cascarano, L. De Caro, C. Giacovazzo, G. Polidori, D. Siliqi and R. Spagna, SIR 2008, 2007 Search PubMed.
- CrystalStructure 4.1: Crystal Structure Analysis Package, Rigaku Corporation, Tokyo 196-8666, Japan, 2000–2014 Search PubMed.
- SHELX97: G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnotes |
| † This paper is dedicated to Herbert C. Brown Distinguished Professor Ei-ichi Negishi at Purdue University on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: The NMR spectra for the products, details on X-ray diffraction analyses. CCDC 1023319, 1023317, 1023315 and 1051704. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00072f |
| This journal is © the Partner Organisations 2015 |