
Reactions of osmapyridinium with terminal alkynes†
Yuanqing
Wei
,
Xiaoxi
Zhou
,
Guangning
Hong
,
Zhixin
Chen
,
Hong
Zhang
* and
Haiping
Xia
*
State Key Laboratory for Physical Chemistry of Solid Surfaces, Department of Chemistry, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, China. E-mail: zh@xmu.edu.cn; hpxia@xmu.edu.cn; Fax: +86-0592-2186628; Tel: +86-0592-2186658
First published on 26th March 2015
Abstract
We have synthesized a new type of ten-membered osmacycles by reaction of osmapyridinium with HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH(OH)R (R = Ph, Et). We propose that these reactions take place initially by coordination of the alkynes, [2 + 2] cycloaddition, subsequent 1,2-hydrogen migration and a final reductive elimination. The reactions with phenylacetylenes do not afford the corresponding derivatives but rather give η4-coordinated cyclopentadiene complexes, which are proposed to derive from a [4 + 2] cycloaddition process. Related reactions of the η4-coordinated cyclopentadiene complexes are also discussed.
CCH(OH)R (R = Ph, Et). We propose that these reactions take place initially by coordination of the alkynes, [2 + 2] cycloaddition, subsequent 1,2-hydrogen migration and a final reductive elimination. The reactions with phenylacetylenes do not afford the corresponding derivatives but rather give η4-coordinated cyclopentadiene complexes, which are proposed to derive from a [4 + 2] cycloaddition process. Related reactions of the η4-coordinated cyclopentadiene complexes are also discussed.
Introduction
The chemistry of metalloaromatic compounds has made significant progress in the past decades.1 The incorporation of a transition metal fragment into conventional aromatic hydrocarbons has recently attracted much attention from both theoretical and experimental chemists.2,3 Whereas the synthesis of a variety of metallobenzene derivatives has been reported and allowed further insight into their reactivities,2 reports on the isolation of analogues containing nitrogen atoms, e.g. metallopyridines, remain limited.4 The first example of metallopyridines is tantalopyridine, which is significant in favoring the localized imido form.4e,f Another example of metallopyridines are osmapyridines with relatively delocalized structures.4a,d It is therefore not surprising that, so far, only the nucleophilic addition reactions4d and the ligand substitution reactions4b of delocalized metallopyridines have been reported.As we were interested in the chemistry of metallacycles, we have studied the reactions of osmafuran with PhCCH and HCCCH(OH)Ph.5 As shown in Chart 1, the reactions yielded nine-membered osmacycles, which might be formed through a [2 + 2] cycloaddition process relevant to olefin metathesis and alkyne polymerization. Another related approach is the investigation of the ruthenium vinyl carbene,6 which can react with propargyl alcohols to produce a series of ten-membered η2-olefine coordinated ruthenacycles (Chart 1(c)). The proposed mechanism for the reaction involves a regioselective [2 + 2] cycloaddition and 1,2-migration process.6a In this paper, we report the reactions of osmapyridinium with terminal alkynes to generate new osmacycles.
 | ||
| Chart 1 (a) Reaction of osmafuran with PhCCH; (b) reaction of osmafuran with HCCCH(OH)Ph; (c) reactions of ruthenium vinyl carbene with propargyl alcohols. | ||
Results and discussion
Reactions of osmapyridinium with HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) CCH(OH)R
CCH(OH)R
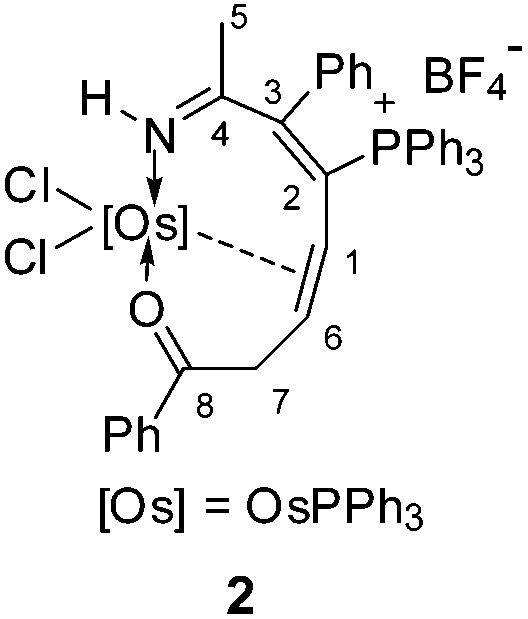
Treatment of osmapyridinium 14d with excess HCCCH(OH)Ph in dichloromethane under reflux led to the formation of complex 2 (Scheme 1). 2 can be isolated as a purple solid in 68% yield, which was characterized by NMR spectroscopy and elemental analysis, and the structure was further confirmed by single-crystal X-ray diffraction.
 | ||
| Scheme 1 Reactions of 1 with HCCCH(OH)R. | ||
The crystallographic details of complex 2 are given in Table 1. As shown in Fig. 1, 2 contains a distorted ten-membered metallacycle (N1, O1, C1, C2, C3, C4, C6, C7, C8, Os1). The osmium centre exhibits a somewhat irregular octahedral geometry, which may be attributed to the coordination of the double bond to the metal centre. The C–C distances within the osmacycle are similar to those in the ten-membered ruthenacycles (Chart 1(c)).6a Consistent with the X-ray structure, the 1H NMR (CD2Cl2) spectrum shows the signals of the coordinated double bond at 5.9 (C1H) and 5.2 (C6H) ppm. The remaining 1H signals of the metallacycle are observed at 10.6 (NH), 3.5 (C7H), and 0.9 ppm (C7H). With the aid of 1H–13C HSQC, 1H–13C HMBC and 13C-dept 135 spectra, the seven carbon signals of the metallacycle are observed at δ = 219.8 (C8), 166.6 (C4), 156.5 (C3), 124.5 (C2), 61.9 (C6), 52.7 (C1), and 43.7 (C7) ppm in the 13C{1H} NMR spectrum. The 31P NMR spectrum shows two signals at 19.7 and −9.5 ppm for CPPh3 and OsPPh3, respectively.
 | ||
| Fig. 1 X-ray structure of complex 2 (ellipsoids at the 50% probability level). Some of the hydrogen atoms, phenyl groups and the counteranion are omitted for clarity. Selected bond lengths (Å) and angles (°): Os1–N1 2.012(7), Os1–O1 2.190(5), N1–C4 1.282(11), C3–C4 1.473(12), C2–C3 1.342(11), C1–C2 1.506(11), C1–C6 1.416(11), C6–C7 1.505(11), C7–C8 1.515(11), C8–O1 1.216(9), C4–C5 1.523(12); O1–Os1–N1 82.6(2), C4–N1–Os1 135.4(6), C3–C4–N1 121.8(7), C2–C3–C4 120.7(7), C1–C2–C3 125.8(7), C2–C1–C6 122.6(7), C1–C6–C7 124.7(7), C6–C7–C8 111.7(7), C7–C8–O1 119.2(7), C8–O1–Os1 116.4(5). | ||
| 2·2H2O·0.25CHCl3 | 5·2CH2Cl2 | 6·CH2Cl2 | 8·2CH2Cl2 | |
|---|---|---|---|---|
| Formula | C56.25H52.25BCl2.75F4NO3OsP2 | C58H52BCl6F4NOOsP2 | C52H55BCl4F4N3OsP | C50H43BCl5F4N3OOsP |
| M r | 1226.68 | 1330.65 | 1171.77 | 1187.10 |
| crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| space group | P21/c | P21/n | Cc | P21/c |
| a [Å] | 11.9299(3) | 16.5731(8) | 19.3445(9) | 12.0746(3) |
| b [Å] | 14.1946(3) | 19.6848(7) | 16.8974(6) | 18.8448(3) |
| c [Å] | 33.0235(7) | 18.1839(9) | 16.1902(7) | 20.9677(4) |
| α [°] | 90 | 90 | 90 | 90 |
| β [°] | 98.5760(10) | 112.182(2) | 111.0040(10) | 90.0230(10) |
| γ [°] | 90 | 90 | 90 | 90 |
| V [Å3] | 5529.7(2) | 5493.2(4) | 4940.5(4) | 4771.06(17) |
| Z | 4 | 4 | 4 | 4 |
| ρ calcd [g cm−3] | 1.473 | 1.609 | 1.574 | 1.653 |
| μ [mm−1] | 2.553 | 2.727 | 2.884 | 3.044 |
| F(000) | 2458 | 2656 | 2352 | 2356 |
| crystal size [mm3] | 0.18 × 0.12 × 0.10 | 0.30 × 0.10 × 0.10 | 1.00 × 0.60 × 0.40 | 0.80 × 0.50 × 0.30 |
| θ Range [°] | 3.08 to 25.00 | 3.02 to 27.48 | 3.25 to 24.99 | 3.11 to 27.48 |
| Reflns collected | 62![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 557 557 |
38256 |
18915 |
70908 |
| Independent reflns | 9691 | 9642 | 7754 | 10901 |
| Observed reflns [I ≥ 2σ(I)] | 8011 | 7791 | 6954 | 8348 |
| Data/restraints/params | 9691/6/667 | 9642/0/669 | 7754/47/596 | 10901/0/598 |
| GOF on F2 | 1.044 | 1.084 | 1.091 | 1.170 |
| R 1/wR2 [I ≥ 2σ(I)]] | 0.0566/0.1635 | 0.0643/0.1710 | 0.0344/0.0737 | 0.0526/0.1396 |
| R 1/wR2 (all data) | 0.0694/0.1720 | 0.0779/0.1873 | 0.0439/0.0858 | 0.0723/0.1954 |
| Largest peak/hole [e Å−3] | 2.14/−1.30 | 4.64/−2.06 | 1.43/−1.15 | 2.13/−2.64 |
Reaction of osmapyridinium 1 with HCCCH(OH)Et was also investigated. As shown in Scheme 1, 1 reacted with HCCCH(OH)Et to produce insertion product 3. 3 has been characterized by NMR spectroscopy and elemental analysis. The structure of 3 can be deduced easily, as its NMR data are similar to those of complex 2 (Table 2). The resonances of the osmacycle in the 1H NMR spectrum (10.5 (N1H), 6.0 (C1H), 5.1 (C6H), 2.8 (C7H), and 0.7 (C7H) ppm) and 13C{1H} NMR spectrum (236.5 (C8), 167.4 (C4), 158.1 (C3), 124.7 (C2), 62.0 (C6), 53.1 (C1), and 48.8 (C7) ppm) are very close to those observed for complex 2.
| Compound | 2 | 3 | |
|---|---|---|---|
| δ (1H) (ppm) | NH | 10.6 | 10.5 |
| H1 | 5.9 | 6.0 | |
| H6 | 5.2 | 5.1 | |
| H7 | 3.5 | 2.8 | |
| 0.9 | 0.7 | ||
| δ (13C) (ppm) | C4 | 166.6 | 167.4 |
| C3 | 156.5 | 158.1 | |
| C2 | 124.5 | 124.7 | |
| C1 | 52.7 | 53.1 | |
| C6 | 61.9 | 62.0 | |
| C7 | 43.7 | 48.8 | |
| C8 | 219.8 | 236.5 | |
Similar to our previous reported mechanism for the formation of ten-membered ruthenacycles from ruthenium vinyl carbene,6a the formation of osmacycles 2 and 3 may also involve a [2 + 2] cycloaddition and 1,2-migration process. As shown in Scheme 2, coordination of the terminal alkyne to the metal center in 1 accompanied by the dissociation of the PPh3 ligand could generate π-alkyne intermediate A. Then, [2 + 2] cycloaddition of the alkyne with the Os![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in A may lead to the formation of the metallacyclobutene intermediate B, which could undergo cycloreversion to form the nine-membered intermediate C. The following activation of hydroxyl in C could produce the hydride intermediate D. Subsequent 1,2-hydrogen migration, reductive elimination, and coordination of the carbonyl group to metal center could yield the final metallacycle 2/3. It is also possible that the proton on the oxygen may transfer to the carbene carbon without the intermediacy of the Os center. The mechanism shown in Scheme 2 is similar to that for our ten-membered ruthenacycle synthesis from ruthenium vinyl carbene and propargyl alcohols as demonstrated by deuterium labeling experiments.6a In this context, it should be mentioned that similar [2 + 2] cycloadditions of alkynes with carbene complexes have been described in literature.7
C in A may lead to the formation of the metallacyclobutene intermediate B, which could undergo cycloreversion to form the nine-membered intermediate C. The following activation of hydroxyl in C could produce the hydride intermediate D. Subsequent 1,2-hydrogen migration, reductive elimination, and coordination of the carbonyl group to metal center could yield the final metallacycle 2/3. It is also possible that the proton on the oxygen may transfer to the carbene carbon without the intermediacy of the Os center. The mechanism shown in Scheme 2 is similar to that for our ten-membered ruthenacycle synthesis from ruthenium vinyl carbene and propargyl alcohols as demonstrated by deuterium labeling experiments.6a In this context, it should be mentioned that similar [2 + 2] cycloadditions of alkynes with carbene complexes have been described in literature.7
 | ||
| Scheme 2 Proposed mechanism of the reaction of 1 with HCCCH(OH)R. | ||
Reactions of osmapyridinium with phenylacetylenes
To further study the reactions of osmapyridinium complex 1 with terminal alkynes, we also investigated its reactivity with phenylacetylenes. As shown in Scheme 3, 1 can react with phenylacetylene to produce complex 4 as a yellow solid in 92% yield. Similarly, complex 5 was obtained in 91% yield from the reaction of 1 with a substituted phenylacetylene, i.e. 1-ethynyl-4-methoxybenzene (Scheme 3). | ||
| Scheme 3 Reactions of 1 with phenylacetylenes. | ||
These two complexes have been characterized by NMR spectroscopy and elemental analysis. As shown in Table 3, the complexes 4 and 5 must have similar structures as indicated by the similarity in their NMR spectroscopic data. Fortunately, we were able to obtain a single crystal for 5, enabling determination of its solid-state structure. The crystallographic details for 5 are given in Table 1. As shown in Fig. 2, 5 contains a substituted cyclopentadiene unit, which is η4-coordinated to the metal centre. The phenyl group and the imido group are located on the sp3 carbon of the cyclopentadiene unit.
 | ||
| Fig. 2 X-ray structure of complex 5 (only the R enantiomer is shown, ellipsoids at the 50% probability level). Some of the hydrogen atoms, phenyl groups and the counteranion are omitted for clarity. Selected bond distances (Å) and angles (°): Os1–N1 2.054(7), Os1–C1 2.206(7), Os1–C2 2.264(7), Os1–C6 2.190(8), Os1–C7 2.307(7), C1–C2 1.395(10), C2–C3 1.595(11), C3–C7 1.577(10), C6–C7 1.419(10), C1–C6 1.424(11), N1–C4 1.258(10), C3–C4 1.520(11), C2–P2 1.805(8); C1–C2–C3 110.1(7), C2–C3–C7 95.4(5), C3–C7–C6 110.1(6), C7–C6–C1 109.1(6), C6–C1–C2 109.1(7), C4–N1–Os1 118.5(6), C3–C4–N1 108.2(7), C3–C4–C5 128.6(7), C2–C3–C4 107.3(6), C7–C3–C4 104.9(6). | ||
| Compound | δ (1H) (ppm) | δ (13C) (ppm) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| NH | H1 | H6 | C4 | C3 | C2 | C1 | C6 | C7 | |
| 4 | 8.3 | 6.8 | 6.9 | 191.0 | 88.3 | 78.6 | 118.2 | 83.4 | 102.9 |
| 5 | 8.3 | 6.8 | 6.7 | 190.7 | 88.3 | 77.6 | 117.4 | 82.1 | 105.6 |
| 6 | 11.1 | 8.2 | 6.5 | 180.3 | 81.9 | 105.3 | 170.4 | 59.2 | 96.7 |
| 7 | 11.1 | 8.2 | 6.5 | 180.2 | 82.0 | 104.9 | 170.6 | 59.5 | 97.1 |
| 8 | 12.2 | 6.1 | 4.7 | 179.6 | 80.3 | 76.3 | 135.4 | 68.9 | 133.8 |
Based on the characterized structures, we postulate the reaction mechanism shown in Scheme 4 for the reaction of 1 with phenylacetylenes. The PPh3 ligand in 1 could initially be displaced by phenylacetylene to give the π-alkyne complex F. Subsequent [4 + 2] cycloaddition of the osmium vinyl carbene fragment with the alkyne in F may lead to the formation of the metallacyclohexadiene intermediate G. Finally, G could undergo reductive elimination to generate complex 4/5 in which the cyclopentadiene is coordinated to the metal center as a ligand. It is interesting to note that, in our previous report, the osmafuran only underwent a head-to-tail double insertion of PhCCH to generate nine-membered osmacycles (Chart 1(a)).5 We speculated that the difference may be attributed to the delocalized structure of the osmapyridinium 1, which only shows a weak alternating double/single bond character around the metallacycle. A similar [4 + 2] cycloaddition process has been previously proposed in the reactions of metallobenzenes with unsaturated substrates8 and the reactions of alkenyl carbene complexes with alkynes or alkenes.9 In the case of osmapyridinium 1 with HCCCH(OH)R, the [2 + 2] cycloaddition may be aided by the hydroxyl group, which helps stabilize the 16-electron intermediate.
 | ||
| Scheme 4 Proposed mechanism of the reactions of 1 with phenylacetylenes. | ||
Reactions of 4/5 with cyclohexyl isocyanide and 2,2′-bipyridine
The osmium complexes 4 and 5 are reactive towards strong ligands, such as isocyanides and bipyridines. As shown in Scheme 5, reactions of 4 and 5 with cyclohexyl isocyanide produce the ligand substitution products 6 and 7, respectively. The structures of 6 and 7 can be assigned based on the NMR data. In particular, the NMR signals associated with the metallacycle are similar to those of the analogous cyclopentadiene complexes 4 and 5 (see Table 3). The different coordination mode of the substituted cyclopentadiene unit in 6 and 7 is supported by the chemical shifts of C1 and C2 in the 13C NMR spectrum. | ||
| Scheme 5 Reactions of 4/5 with cyclohexyl isocyanide. | ||
The structure of complex 6 was further confirmed by X-ray diffraction analysis (Fig. 3). As shown in Fig. 3 and Table 4, the structural parameters of complex 6 are similar to those of 5. The two cyclohexyl isocyanide ligands are mutually trans, and the substituted cyclopentadiene fragment in 6 is η2-coordinated to the metal centre. We think that the steric effect of the bulky phosphonium group would account for the observed regioselectivity.
 | ||
| Fig. 3 X-ray structure of complex 6 (only the R enantiomer is shown, ellipsoids at the 50% probability level). Some of the hydrogen atoms, phenyl groups and the counteranion are omitted for clarity. | ||
| 6 | 8 | |
|---|---|---|
| Bond distances (Å) | ||
| Os1–N1 | 2.020(6) | 2.022(5) |
| N1–C4 | 1.280(9) | 1.290(7) |
| C3–C4 | 1.546(10) | 1.532(8) |
| C1–C2 | 1.360(10) | 1.455(7) |
| C2–C3 | 1.552(10) | 1.593(7) |
| C3–C7 | 1.602(9) | 1.535(8) |
| C6–C7 | 1.412(10) | 1.323(9) |
| C1–C6 | 1.454(9) | 1.459(8) |
| Bond angles (°) | ||
| Os1–N1–C4 | 125.6(5) | 126.5(4) |
| N1–C4–C3 | 113.7(6) | 113.1(5) |
| C4–C3–C2 | 103.4(5) | 106.6(4) |
| C4–C3–C7 | 108.0(6) | 103.4(5) |
| C1–C2–C3 | 109.9(6) | 106.0(4) |
| C2–C3–C7 | 100.6(5) | 101.5(4) |
| C3–C7–C6 | 108.2(6) | 111.7(6) |
| C1–C6–C7 | 108.8(6) | 111.9(5) |
| C2–C1–C6 | 112.3(6) | 108.7(5) |
The reaction of 5 with 2,2′-bipyridine was also examined. Compound 8 could be readily obtained in 75% isolated yield by the reaction of 5 with 2,2′-bipyridine (Scheme 6). The complex has been characterized by NMR spectroscopy and elemental analysis. The structures of complexes 8 were assigned based on the fact that the NMR data associated with the complex are similar to those of 4, 5, 6 and 7, as illustrated by the data shown in Table 3.
 | ||
| Scheme 6 Reactions of 5 with 2,2′-bipyridine. | ||
The structure of 8 has been confirmed by X-ray diffraction (Fig. 4). Like 6, the cyclopentadiene ligand in 8 is also η2-coordinated to osmium. The complex contains the 2,2′-bipyridine in the equatorial plane, and one of the phenyl groups of the phosphonium ligand bonded to the metal center which may derive from the C–H activation process. We have reported a similar oxidative addition of the phenyl C–H bond of PPh3 which could give rise to tethered complexes.10 In this case, the basicity of the 2,2′-bipyridine might facilitate the C–H activation of the phosphonium ligand. Thus, the reaction of 5 with pyridine under the above reaction conditions resulted in the formation of a similar tethered complex, as suggested by in situ NMR.
 | ||
| Fig. 4 X-ray structure of complex 8 (only the R enantiomer is shown, ellipsoids at the 50% probability level). Some of the hydrogen atoms, phenyl groups and the counteranion are omitted for clarity. | ||
Conclusions
The reactions of osmapyridinium with terminal alkynes were studied, which led to the formation of new osmium complexes. For propargyl alcohols, the [2 + 2] cycloaddition and 1,2-hydrogen migration process was proposed for the formation of the final ten-membered osmacycles. For phenylacetylenes, η4-coordinated cyclopentadiene complexes can be obtained, which may be attributed to a [4 + 2] cycloaddition process.Experimental
General comments
All manipulations were carried out at room temperature under a nitrogen atmosphere using standard Schlenk techniques, unless otherwise stated. Solvents were distilled under nitrogen from sodium benzophenone (diethyl ether) or calcium hydride (dichloromethane). Column chromatography was performed on neutral alumina gel (200–300 mesh) or silica gel (200–300 mesh). NMR experiments were performed on a Bruker AV-400 spectrometer (1H 400.1 MHz; 13C 101.6 MHz; 31P 162.0 MHz) at room temperature unless otherwise stated. 1H and 13C NMR chemical shifts are relative to TMS, and 31P NMR chemical shifts are relative to 85% H3PO4. Elemental analysis data were obtained on an Elementar Analysensystem GmbH Vario EL III instrument.



 NCH), 2.0 (s, 3 H, H5), 2.0–1.0 ppm (20 H, other alkyl carbon atoms). 31P{1H} NMR (162.0 MHz, CD2Cl2): δ = 18.1 ppm (s, CPPh3). 13C{1H} NMR (101.6 MHz, CD2Cl2, plus 1H–13C HSQC and 1H–13C HMBC and 13C-dept 135): δ = 180.3 (s, C4), 170.4 (d, 2J(P, C) = 11.3 Hz, C1), 140.5–116.8 (other aromatic carbon atoms), 105.3 (d, 1J(P, C) = 85.8 Hz, C2), 96.7 (d, 3J(P, C) = 6.9 Hz, C7), 81.9 (d, 2J(P, C) = 8.1 Hz, C3), 59.2 (d, 3J(P, C) = 16.2 Hz, C6), 54.3 (s, CNCH), 52.8 (s, CNCH), 31.9–21.3 (other alkyl carbon atoms), 25.6 ppm (s, C5). Elemental analysis calcd (%) for C51H52Cl2PBF4N3Os: C, 56.41; H, 4.83; N, 3.87. Found: C, 56.80; H, 5.10; N, 3.76.
NCH), 2.0 (s, 3 H, H5), 2.0–1.0 ppm (20 H, other alkyl carbon atoms). 31P{1H} NMR (162.0 MHz, CD2Cl2): δ = 18.1 ppm (s, CPPh3). 13C{1H} NMR (101.6 MHz, CD2Cl2, plus 1H–13C HSQC and 1H–13C HMBC and 13C-dept 135): δ = 180.3 (s, C4), 170.4 (d, 2J(P, C) = 11.3 Hz, C1), 140.5–116.8 (other aromatic carbon atoms), 105.3 (d, 1J(P, C) = 85.8 Hz, C2), 96.7 (d, 3J(P, C) = 6.9 Hz, C7), 81.9 (d, 2J(P, C) = 8.1 Hz, C3), 59.2 (d, 3J(P, C) = 16.2 Hz, C6), 54.3 (s, CNCH), 52.8 (s, CNCH), 31.9–21.3 (other alkyl carbon atoms), 25.6 ppm (s, C5). Elemental analysis calcd (%) for C51H52Cl2PBF4N3Os: C, 56.41; H, 4.83; N, 3.87. Found: C, 56.80; H, 5.10; N, 3.76.
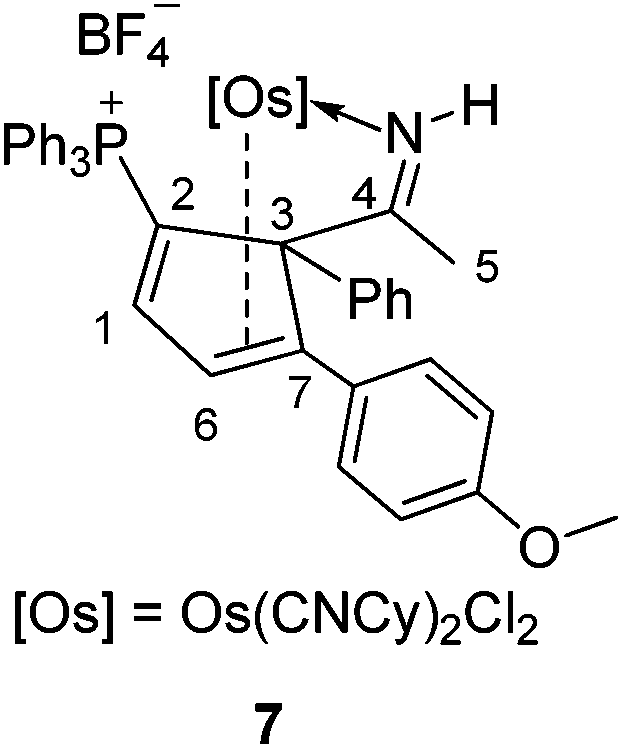 NCH), 3.6 (s, 3 H, OCH3), 2.0 (s, 3 H, H5), 1.9–1.1 ppm (20 H, other alkyl carbon atoms). 31P{1H} NMR (162.0 MHz, CD2Cl2): δ = 18.0 ppm (s, CPPh3). 13C{1H} NMR (101.6 MHz, CD2Cl2, plus 1H–13C HSQC and 1H–13C HMBC and 13C-dept 135): δ = 180.2 (s, C4), 170.6 (d, 2J(P, C) = 11.1 Hz, C1), 157.2 (s, CN), 134.8–111.5 (other aromatic carbon atoms), 104.9 (d, 1J(P, C) = 85.6 Hz, C2), 97.1 (d, 3J(P, C) = 7.5 Hz, C7), 82.0 (d, 2J(P, C) = 7.5 Hz, C3), 59.5 (d, 3J(P, C) = 16.2 Hz, C6), 54.3 (s, OCH3), 54.3 (s, CNCH), 31.9–21.3 (other alkyl carbon atoms), 25.6 ppm (s, C5). Elemental analysis calcd (%) for C52H55Cl2PBF4N3OOs: C, 55.92; H, 4.96; N, 3.76. Found: C, 55.99; H, 4.98; N, 3.48.
NCH), 3.6 (s, 3 H, OCH3), 2.0 (s, 3 H, H5), 1.9–1.1 ppm (20 H, other alkyl carbon atoms). 31P{1H} NMR (162.0 MHz, CD2Cl2): δ = 18.0 ppm (s, CPPh3). 13C{1H} NMR (101.6 MHz, CD2Cl2, plus 1H–13C HSQC and 1H–13C HMBC and 13C-dept 135): δ = 180.2 (s, C4), 170.6 (d, 2J(P, C) = 11.1 Hz, C1), 157.2 (s, CN), 134.8–111.5 (other aromatic carbon atoms), 104.9 (d, 1J(P, C) = 85.6 Hz, C2), 97.1 (d, 3J(P, C) = 7.5 Hz, C7), 82.0 (d, 2J(P, C) = 7.5 Hz, C3), 59.5 (d, 3J(P, C) = 16.2 Hz, C6), 54.3 (s, OCH3), 54.3 (s, CNCH), 31.9–21.3 (other alkyl carbon atoms), 25.6 ppm (s, C5). Elemental analysis calcd (%) for C52H55Cl2PBF4N3OOs: C, 55.92; H, 4.96; N, 3.76. Found: C, 55.99; H, 4.98; N, 3.48.

Crystallographic details
Single-crystal X-ray diffraction data were collected on an Oxford Gemini S Ultra CCD Area Detector or a Rigaku R-AXIS SPIDER IP CCD Area Detector with graphite-monochromated MoKa radiation (λ = 0.71073 Å). All of the data were corrected for absorption effects using the multi-scan technique. The structures were solved by direct methods, expanded by difference Fourier syntheses and refined by full matrix least-squares on F2 using Bruker SHELXTL (Version 6.10) program package. Non-H atoms were refined anisotropically unless otherwise stated. Hydrogen atoms were introduced at their geometric positions and refined as riding atoms unless otherwise stated. For complexes 5, 6 and 8, the crystal suitable for X-ray diffraction was grown from a CH2Cl2 solution layered with hexane. For complex 2, the crystal suitable for X-ray diffraction was grown from a CHCl3 solution layered with hexane. Solvent molecules CHCl3 in 2 are disordered and were refined with suitable restraints. Isocyanocyclohexane ligand molecules in 6 were refined isotropically using fixed C–C or C–N length. CCDC-1038534 (2), CCDC-1038533 (5), CCDC-1038537 (6), CCDC-1038535 (8), please see the supplementary crystallographic data for this paper.Acknowledgements
This research was supported by the National Natural Science Foundation of China (21272193, 21490573) and the program for Changjiang Scholars and Innovative Research Team in University.Notes and references
- For recent reviews on the chemistry of metalloaromatics, see: (a) B. J. Frogley and L. J. Wright, Coord. Chem. Rev., 2014, 270–271, 151–166 CrossRef CAS PubMed; (b) X.-Y. Cao, Q. Zhao, Z. Lin and H. Xia, Acc. Chem. Res., 2014, 47, 341–354 CrossRef CAS PubMed; (c) C. Zhu, X. Cao and H. Xia, Chin. J. Org. Chem., 2013, 33, 657–662 CrossRef CAS; (d) G. Jia, Organometallics, 2013, 32, 6852–6866 CrossRef CAS; (e) J. Chen and G. Jia, Coord. Chem. Rev., 2013, 257, 2491–2521 CrossRef CAS PubMed; (f) J. Chen, G. He and G. Jia, Chin. J. Org. Chem., 2013, 33, 792–798 CrossRef CAS; (g) A. F. Dalebrook and L. J. Wright, Adv. Organomet. Chem., 2012, 60, 93–177 CrossRef CAS; (h) M. Paneque, M. L. Poveda and N. Rendón, Eur. J. Inorg. Chem., 2011, 19–33 CrossRef CAS; (i) A. T. Balaban, Top. Heterocycl. Chem., 2009, 19, 203–246 CAS; (j) G. Jia, Coord. Chem. Rev., 2007, 251, 2167–2187 CrossRef CAS PubMed; (k) J. R. Bleeke, Acc. Chem. Res., 2007, 40, 1035–1047 CrossRef CAS PubMed; (l) L. J. Wright, Dalton Trans., 2006, 1821–1827 RSC; (m) C. W. Landorf and M. M. Haley, Angew. Chem., Int. Ed., 2006, 45, 3914–3936 CrossRef CAS PubMed; (n) G. Jia, Acc. Chem. Res., 2004, 37, 479–486 CrossRef CAS PubMed; (o) G. He, H. Xia and G. Jia, Chin. Sci. Bull., 2004, 49, 1543–1553 CAS; (p) J. R. Bleeke, Chem. Rev., 2001, 101, 1205–1227 CrossRef CAS PubMed.
- For very recent reports on metallobenzenes: (a) F. Han, J. Li, H. Zhang, T. Wang, Z. Lin and H. Xia, Chem. – Eur. J., 2015, 21, 565–567 CrossRef CAS PubMed; (b) R. Lin, K.-H. Lee, K. C. Poon, H. H. Y. Sung, I. D. Williams, Z. Lin and G. Jia, Chemistry, 2014, 20, 14885–14899 CrossRef CAS PubMed; (c) F. Han, T. Wang, J. Li, H. Zhang and H. Xia, Chem. – Eur. J., 2014, 20, 4363–4372 CrossRef CAS PubMed; (d) Q. Zhao, X.-Y. Cao, T. B. Wen and H. Xia, Chem. – Asian J., 2013, 8, 269–275 CrossRef CAS PubMed; (e) T. Wang, J. Zhu, F. Han, C. Zhou, H. Chen, H. Zhang and H. Xia, Angew. Chem., Int. Ed., 2013, 52, 13361–13364 CrossRef CAS PubMed; (f) T. Wang, H. Zhang, F. Han, L. Long, Z. Lin and H. Xia, Chem. – Eur. J., 2013, 19, 10982–10991 CrossRef CAS PubMed; (g) T. Wang, H. Zhang, F. Han, L. Long, Z. Lin and H. Xia, Angew. Chem., Int. Ed., 2013, 52, 9251–9255 CrossRef CAS PubMed; (h) Á. Vivancos, M. Paneque, M. L. Poveda and E. Álvarez, Angew. Chem., Int. Ed., 2013, 52, 10068–10071 CrossRef PubMed; (i) J. Chen, C. Zhang, T. Xie, T. B. Wen, H. Zhang and H. Xia, Organometallics, 2013, 32, 3993–4001 CrossRef CAS; (j) Q. Zhao, J. Zhu, Z.-A. Huang, X.-Y. Cao and H. Xia, Chem. – Eur. J., 2012, 18, 11597–11603 CrossRef CAS PubMed; (k) R. Lin, J. Zhao, H. Chen, H. Zhang and H. Xia, Chem. – Asian J., 2012, 7, 1915–1924 CrossRef CAS PubMed; (l) M. Hernández-Juárez, V. Salazar, E. V. García-Báez, I. I. Padilla-Martínez, H. Höpfl and M. d. J. Rosales-Hoz, Organometallics, 2012, 31, 5438–5451 CrossRef; (m) Q. Zhao, L. Gong, C. Xu, J. Zhu, X. He and H. Xia, Angew. Chem., Int. Ed., 2011, 50, 1354–1358 CrossRef CAS PubMed; (n) C. Shi, T. Guo, K. C. Poon, Z. Lin and G. Jia, Dalton Trans., 2011, 40, 11315–11320 RSC; (o) R. Lin, H. Zhang, S. Li, J. Wang and H. Xia, Chem. – Eur. J., 2011, 17, 4223–4231 CrossRef CAS PubMed; (p) R. Lin, H. Zhang, S. Li, L. Chen, W. Zhang, T. B. Wen, H. Zhang and H. Xia, Chem. – Eur. J., 2011, 17, 2420–2427 CrossRef CAS PubMed; (q) H. Zhang, R. Lin, G. Hong, T. Wang, T. B. Wen and H. Xia, Chem. – Eur. J., 2010, 16, 6999–7007 CrossRef CAS PubMed; (r) K. C. Poon, L. Liu, T. Guo, J. Li, H. H. Y. Sung, I. D. Williams, Z. Lin and G. Jia, Angew. Chem., Int. Ed., 2010, 49, 2759–2762 CrossRef CAS PubMed; (s) P. M. Johns, W. R. Roper, S. D. Woodgate and L. J. Wright, Organometallics, 2010, 29, 5358–5365 CrossRef CAS; (t) J. Huang, R. Lin, L. Wu, Q. Zhao, C. Zhu, T. B. Wen and H. Xia, Organometallics, 2010, 29, 2916–2925 CrossRef CAS; (u) G. R. Clark, L. A. Ferguson, A. E. McIntosh, T. Söhnel and L. J. Wright, J. Am. Chem. Soc., 2010, 132, 13443–13452 CrossRef CAS PubMed.
- For very recent reports on other metalloaromatics: (a) Y. Huang and J. Zhu, Chem. – Asian J., 2015, 10, 405–410 CrossRef CAS PubMed; (b) Y. Zeng, H. Feng, R. B. King and H. F. Schaefer III, Organometallics, 2014, 33, 7193–7198 CrossRef CAS; (c) C. Huang, Y. Hao, Y. Zhao and J. Zhu, Organometallics, 2014, 33, 817–822 CrossRef CAS; (d) J. Fan, X. Wang and J. Zhu, Organometallics, 2014, 33, 2336–2340 CrossRef CAS; (e) M. El-Hamdi, O. El Bakouri, E. Farri, P. Salvador, B. A. Abdelouahid, M. S. El Begrani, J. Poater and M. Solà, Organometallics, 2013, 32, 4892–4903 CrossRef CAS; (f) R. W. A. Havenith, F. De Proft, L. W. Jenneskens and P. W. Fowler, Phys. Chem. Chem. Phys., 2012, 14, 9897–9905 RSC; (g) M. Lin, P. Li and Z. Cao, J. Theor. Comput. Chem., 2011, 10, 861–874 CrossRef CAS; (h) M. Mauksch and S. B. Tsogoeva, Chem. – Eur. J., 2010, 16, 7843–7851 CrossRef CAS PubMed; (i) Z. Lin, Acc. Chem. Res., 2010, 43, 602–611 CrossRef CAS PubMed.
- Metallopyridines: (a) T. Wang, H. Zhang, F. Han, R. Lin, Z. Lin and H. Xia, Angew. Chem., Int. Ed., 2012, 51, 9838–9841 CrossRef CAS PubMed; (b) B. Liu, Q. Zhao, H. Wang, J. Chen, X. Cao, Z. Cao and H. Xia, Chin. J. Chem., 2012, 30, 2158–2168 CrossRef CAS; (c) H. Zhang, R. Lin, G. Hong, T. Wang, T. B. Wen and H. Xia, Chem. – Eur. J., 2010, 16, 6999–7007 CrossRef CAS PubMed; (d) B. Liu, H. Wang, H. Xie, B. Zeng, J. Chen, J. Tao, T. B. Wen, Z. Cao and H. Xia, Angew. Chem., Int. Ed., 2009, 48, 5430–5434 CrossRef CAS PubMed; (e) K. J. Weller, I. Filippov, P. M. Briggs and D. E. Wigley, Organometallics, 1998, 17, 322–329 CrossRef CAS; (f) K. J. Weller, I. Filippov, P. M. Briggs and D. E. Wigley, J. Organomet. Chem., 1997, 528, 225–228 CrossRef CAS.
- Y. Lin, L. Gong, H. Xu, X. He, T. B. Wen and H. Xia, Organometallics, 2009, 28, 1524–1533 CrossRef CAS.
- (a) X. Zhou, C. Zhang, Y. Lin, X. He, Y. Zhang, J. Wang and H. Xia, Org. Chem. Front., 2014, 1, 1077–1082 RSC; (b) C. Zhang, H. Zhang, L. Zhang, T. B. Wen, X. He and H. Xia, Organometallics, 2013, 32, 3738–3743 CrossRef CAS; (c) C. Zhang, H. Zhang, A. Wei, X. He and H. Xia, Acta Chim. Sin., 2013, 71, 1373–1378 CrossRef CAS.
- See for example: (a) K. H. Ng, Y. Li, R. Ganguly and F. Mathey, Organometallics, 2013, 32, 7482–7486 CrossRef CAS; (b) H. Park and T.-L. Choi, J. Am. Chem. Soc., 2012, 134, 7270–7273 CrossRef CAS PubMed; (c) M. Yamaguchi, Y. Arikawa, Y. Nishimura, K. Umakoshi and M. Onishi, Chem. Commun., 2009, 2911–2913 RSC; (d) W. Zhang, J. Yamada and K. Nomura, Organometallics, 2008, 27, 5353–5536 CrossRef CAS.
- (a) V. Jacob, C. W. Landorf, L. N. Zakharov, T. J. R. Weakley and M. M. Haley, Organometallics, 2009, 28, 5183–5190 CrossRef CAS; (b) J. R. Bleeke, Acc. Chem. Res., 2007, 40, 1035–1047 CrossRef CAS PubMed; (c) M. A. Iron, A. C. B. Lucassen, H. Cohen, M. E. van der Boom and J. M. L. Martin, J. Am. Chem. Soc., 2004, 126, 11699–11710 CrossRef CAS PubMed; (d) M. A. Iron, J. M. L. Martin and M. E. van der Boom, J. Am. Chem. Soc., 2003, 125, 11702–11709 CrossRef CAS PubMed.
- (a) J. Barluenga, R. Vicente, L. A. López, E. Rubio, M. Tomás and C. Álvarez-Rúa, J. Am. Chem. Soc., 2004, 126, 470–471 CrossRef CAS PubMed; (b) J. Barluenga, S. López and J. Flórez, Angew. Chem., Int. Ed., 2003, 42, 231–233 CrossRef CAS PubMed; (c) H. Kagoshima, T. Okamura and T. Akiyama, J. Am. Chem. Soc., 2001, 123, 7182–7183 CrossRef CAS; (d) M. Hoffmann, M. Buchert and H.-U. Reißig, Chem. – Eur. J., 1999, 5, 876–882 CrossRef CAS; (e) B. L. Flynn, F. J. Funke, C. C. Silveira and A. d. Meijere, Synlett, 1995, 1007–1010 CrossRef CAS PubMed; (f) B. M. Trost and A. S. K. Hashmi, J. Am. Chem. Soc., 1994, 116, 2183–2184 CrossRef CAS.
- (a) F. Han, T. Wang, J. Li, H. Zhang, L. Zhang, X. He and H. Xia, Organometallics, 2014, 33, 5301–5307 CrossRef CAS; (b) Y. Lin, H. Xu, L. Gong, T. B. Wen, X.-M. He and H. Xia, Organometallics, 2010, 29, 2904–2910 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Material including copies of 1H and 13C NMR spectra of all new products and crystallographic data for 2, 5, 6, 8. CCDC 1038534, 1038533, 1038537 and 1038535. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00052a |
| This journal is © the Partner Organisations 2015 |