DOI:
10.1039/C5QO00050E
(Research Article)
Org. Chem. Front., 2015,
2, 552-559
Development of a functionally separated D–π-A fluorescent dye with a pyrazyl group as an electron-accepting group for dye-sensitized solar cells†
Received
9th February 2015
, Accepted 13th March 2015
First published on 16th March 2015
Abstract
A functionally separated D–π-A dye OUK-3 with a pyrazyl group as an electron-withdrawing anchoring group and a carboxyl group as an additional anchoring group has been newly developed as a photosensitizer for dye-sensitized solar cells. The optical and electrochemical properties, adsorption states on TiO2 nanoparticles, and photovoltaic performances in dye-sensitized solar cells (DSSCs) were investigated. It was found that the maximum adsorption amount of dye adsorbed on the TiO2 electrode for OUK-3 is 3 times as much as that of the D–π-A dye sensitizer OUK-1 with a pyrazyl group as an electron-withdrawing anchoring group. Moreover, this work revealed that the DSSC based on the dye OUK-3 exhibits a higher photovoltage (Voc) value than the DSSC based on the dye OUK-1. On the basis of the FTIR spectra of the dyes adsorbed on TiO2 nanoparticles and the electrochemical impedance spectroscopy (EIS) analysis of DSSCs based on the two dyes OUK-1 and OUK-3, the differences of photovoltaic performances between the two dyes are discussed by taking into account the adsorption states of the dyes adsorbed on the TiO2 surface. This work demonstrates that functionally separated D–π-A dye sensitizers can achieve effective surface coverage of the TiO2 electrode due to their high adsorption ability onto the TiO2 electrode, leading to not only the improvement of light-harvesting efficiency (LHE), but also an increase in the number of injected electrons in the CB of TiO2, which is responsible for the higher Voc value of functionally separated D–π-A dye sensitizers.
Introduction
Dye-sensitized solar cells (DSSCs) employing dye-adsorbed TiO2 electrodes have attracted much attention from chemists, physicists, and engineers because of enormous scientific interest in their construction and operational principles, and low cost of production since Grätzel and co-workers produced high-performance DSSCs based on a Ru-complex dye in 1991.1–10 Much effort in molecular design and development of efficient dye sensitizers have been made to further improve the photovoltaic performances of DSSCs so far. In particular, donor–acceptor π-conjugated (D–π-A) dye sensitizers having both electron-donating (D) and electron-accepting (A) groups linked by π-conjugated bridges would be expected to be one of the most promising classes of organic dye sensitizers, because the wide variety of structures and facile modification provides potential for molecular design, with the introduction of substituents onto the chromophore skeletons allowing easy control not only of their photophysical and electrochemical properties (HOMO and LUMO levels), but also of their stereochemical structures.4–10 Consequently, a new type of D–π-A dye sensitizers with 2-(1,1-dicyanomethylene)rhodanine,11 pyridine,12,13 and 8-hydroxylquinoline14 as an electron-withdrawing anchoring group, as well as conventional D–π-A dye sensitizers with a carboxyl group have been designed and developed so far. Many research groups demonstrated that the conventional D–π-A dye sensitizers with a carboxyl group are adsorbed on the TiO2 film through the bidentate bridging linkage between the carboxyl group of the dye and Brønsted acid sites (surface-bound hydroxyl groups, Ti–OH) on the TiO2 surface.4–10 On the other hand, we have demonstrated that dye sensitizers with a pyridyl group are predominantly adsorbed on the TiO2 through coordinate bonding between the pyridyl group of the dye and the Lewis acid site (exposed Tin+ cations) on the TiO2 surface.12 It was found that D–π-A dye sensitizers with a pyridyl group can inject electrons efficiently to the conduction band (CB) of the TiO2 electrode through coordinate bonding, rather than conventional D–π-A dye sensitizers with carboxyl groups. Recently, DSSCs based on porphyrin dyes and D–π-A dyes with pyridyl groups have been reported by some research groups.13
Moreover, to seek an epoch-making molecular design of D–π-A dye sensitizers for high-performance DSSCs, some research groups designed and synthesized the functionally separated D–π-A dye sensitizers with a pyridyl group,12a,c a cyano group,15,16 a nitro group17 or an aldehyde18 as an electron-accepting group and a carboxyl group as an additional anchoring group. They investigated the effects of the interaction between the electron-accepting group of the dye and the TiO2 surface on the photovoltaic performances of DSSCs. It was revealed that the biggest advantage of the functionally separated D–π-A dye sensitizers is that one can introduce both strong electron-withdrawing groups into the electron acceptor moiety and an additional anchoring group, leading to not only high adsorption ability onto the TiO2 film, but also the bathochromic shift and broadening of the photoabsorption property and efficient electron injection from the dye to the CB of the TiO2 electrode. For example, Sun and co-workers designed and synthesized efficient near infrared D–π-A dye sensitizers, with strong electron-withdrawing groups as the electron acceptors, in which the anchoring group (carboxyl group) in these dyes was separated from the electron acceptor unit (dicyanomethylene).15 They also developed the functionally separated D–π-A dye sensitizer with a nitro group as the electron-accepting group and a carboxyl group as the anchoring group. They demonstrated the effects of the interaction between the nitro group of the dye and the TiO2 surface on the photovoltaic performances and UV-Vis absorption properties.17 Thus, the functionally separated D–π-A dye sensitizers would also be expected to be one of the most promising classes of organic dye sensitizers for DSSCs.
In this work, a functionally separated D–π-A dye sensitizer OUK-3 with a pyrazyl group as an electron-withdrawing anchoring group and a carboxyl group as an additional anchoring group has been newly developed. The optical and electrochemical properties, adsorption states on TiO2 nanoparticles, and photovoltaic performances in dye-sensitized solar cells (DSSCs) were investigated (Scheme 1). It was found that the maximum adsorption amount of dyes adsorbed on the TiO2 electrode for OUK-3 is 3 times as much as that of the D–π-A dye sensitizer OUK-1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19 with a pyrazyl group as an electron-withdrawing anchoring group. Moreover, this work revealed that the DSSC based on the dye OUK-3 exhibits a higher photovoltage (Voc) value than the DSSC based on the dye OUK-1. On the basis of the FTIR spectra of the dyes adsorbed on TiO2 nanoparticles and the electrochemical impedance spectroscopy (EIS) analysis of DSSCs based on the two dyes OUK-1 and OUK-3, the differences of photovoltaic performances between the two dyes are discussed by taking into account the adsorption states of the dyes adsorbed on the TiO2 surface.
19 with a pyrazyl group as an electron-withdrawing anchoring group. Moreover, this work revealed that the DSSC based on the dye OUK-3 exhibits a higher photovoltage (Voc) value than the DSSC based on the dye OUK-1. On the basis of the FTIR spectra of the dyes adsorbed on TiO2 nanoparticles and the electrochemical impedance spectroscopy (EIS) analysis of DSSCs based on the two dyes OUK-1 and OUK-3, the differences of photovoltaic performances between the two dyes are discussed by taking into account the adsorption states of the dyes adsorbed on the TiO2 surface.
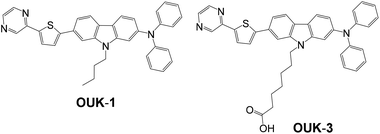 |
| | Scheme 1 Chemical structures of the D–π-A dye sensitizer OUK-1 and the functionally separated D–π-A dye sensitizer OUK-3. | |
Results and discussion
Synthesis
The synthesis of OUK-1 has been reported elsewhere.19 The synthetic pathway of OUK-3 is shown in Scheme 2. The starting material 119 was converted to stannyl compound 2 by treatment with nBuLi and then Me3SnCl. Compound 3 was prepared by the Stille coupling of 2 with 2-iodopyrazine. The reaction of 3 with ethyl 7-bromoheptanoate by using sodium hydride yielded the compound 4, and then the hydrolysis of 4 with a base gave the functionally separated D–π-A dye sensitizer OUK-3.
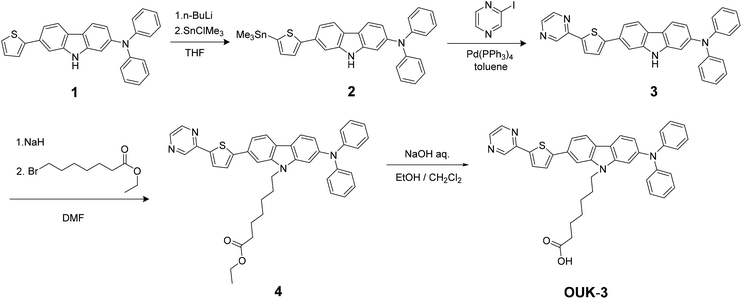 |
| | Scheme 2 Synthetic pathway of the functionally separated D–π-A dye sensitizer OUK-3. | |
Optical properties
The absorption and fluorescence spectra of OUK-3 in 1,4-dioxane are shown in Fig. 1a (see Fig. S1a in ESI† for OUK-1). The two dyes show the absorption maximum (λabsmax) at 402 nm, which is assigned to the intramolecular charge-transfer (ICT) excitation from the electron donor moiety (diphenylamino group) to the electron acceptor moiety (pyrazyl group). The molar extinction coefficient (ε) for the ICT band is 45400 M−1 cm−1 for OUK-1 and 48300 M−1 cm−1 for OUK-3, respectively. The corresponding fluorescence bands for the two dyes occur at around 480 nm, and the fluorescence quantum yield (Φfl) is 0.46 for OUK-1 and 0.48 for OUK-3, respectively (Table 1).
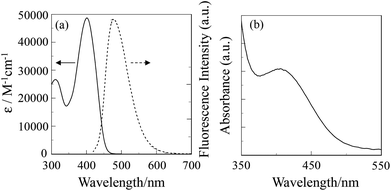 |
| | Fig. 1 Absorption (–) and fluorescence (⋯) spectra of OUK-3 in 1,4-dioxane and (b) absorption spectrum of OUK-3 adsorbed on the TiO2 film. | |
Table 1 Optical and electrochemical data, HOMO and LUMO energy levels, and DSSC performance parameters of OUK-1 and OUK-3
| Dye |
λ absmax/nm (ε M−1 cm−1) |
λ flmaxa/nm (Φf) |
E ox1/2b/V |
HOMOc/V |
LUMOc/V |
Moleculesd/cm−2 |
J sce/mA cm−2 |
V oce/mV |
FFe |
η (%) |
| In 1,4-dioxane. Fluorescence quantum yields (Φf) were determined by using a calibrated integrating sphere system (λex = 402 for OUK-1 and OUK-3). Half-wave potentials for oxidation (Eox1/2) vs. Fc/Fc+ were recorded in the DMF–Bu4NClO4 (0.1 M) solution. vs. Normal hydrogen electrode (NHE). The 9 μm thick TiO2 electrode was immersed into a 1 mM dye solution in THF for 15 hours. The dye-coated film was immersed in a mixed solvent of THF–DMSO–NaOH aq. 1 M (5:4:1), which was used to determine the amount of dye molecules adsorbed onto the film by measuring the absorbance. Under a simulated solar light (AM 1.5, 100 mW cm−2). |
| OUK-1 |
402 (45400) |
478 (0.46) |
0.39 |
1.11 |
−1.68 |
3.0 × 1016 |
2.99 |
448 |
0.67 |
0.89 |
| OUK-3 |
402 (48300) |
477 (0.48) |
0.39 |
1.11 |
−1.69 |
9.6 × 1016 |
4.48 |
550 |
0.64 |
1.58 |
The absorption spectrum of the dye OUK-3 adsorbed on the TiO2 film is shown in Fig. 1b (see Fig. S1b in ESI† for OUK-1). The absorption bands of OUK-1 and OUK-3 adsorbed on the TiO2 film are broadened compared with those in 1,4-dioxane. However, the absorption peak wavelengths of OUK-1 and OUK-3 adsorbed on TiO2 are similar to those in 1,4-dioxane. Thus, this result indicates that the two dyes form weak π-stacked aggregates on the TiO2 surface.
Electrochemical properties
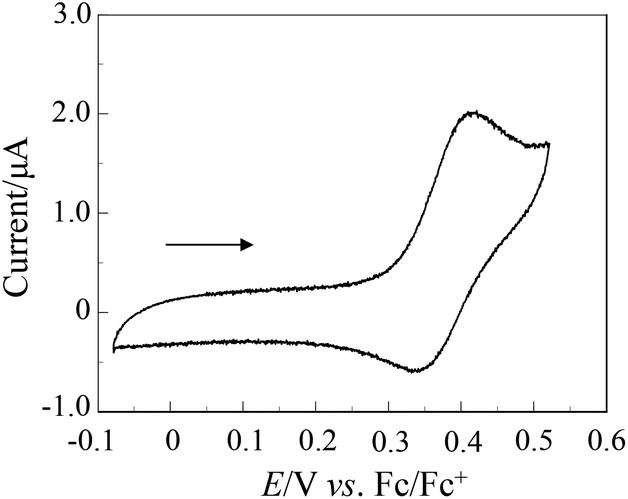
The electrochemical properties of OUK-1 and OUK-3 were determined by cyclic voltammetry (Fig. 2 and Table 1, see Fig. S2 in ESI† for OUK-1). The reversible oxidation waves for the two dyes were observed at 0.42 V vs. ferrocene/ferrocenium (Fc/Fc+). The corresponding reduction waves for the two dyes appeared at 0.35 V, thus showing that the oxidized states of the two dyes are stable. The HOMO energy level was evaluated from the half-wave potential for oxidation (Eox1/2 = 0.39 V for the two dyes). The HOMO energy level was 1.11 V vs. the normal hydrogen electrode (NHE), thus indicating that the HOMO energy level is more positive than the I3–/I– redox potential (0.4 V). This assures efficient regeneration of the oxidized dyes by electron transfer from the I3–/I– in the electrolyte. The LUMO energy level was estimated from the Eox1/2 and an intersection of the absorption and fluorescence spectra (445 nm; 2.79 eV for OUK-1 and 443 nm; 2.80 eV for OUK-3). The LUMO energy levels of OUK-1 and OUK-3 were −1.68 and −1.69 V, respectively. Evidently, the LUMO energy levels of OUK-1 and OUK-3 are higher than the energy level (Ecb) of the CB of TiO2 (–0.5 V), suggesting that an electron injection to the CB of TiO2 is thermodynamically feasible. Accordingly, the optical and electrochemical properties of OUK-1 and OUK-3 resemble each other very closely, showing that the effect of N-alkylation of the carbazole ring on the photophysical and electrochemical properties is negligible.
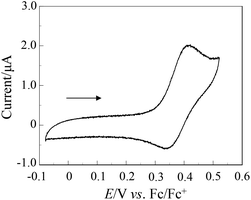 |
| | Fig. 2 Cyclic voltammogram of OUK-1 (0.1 mM) in DMF containing 0.1 M Bu4NClO4 at a scan rate of 10 mV s−1. The arrow denotes the direction of the potential scan. | |
Theoretical calculations
To examine the electronic structures of OUK-1 and OUK-3, the molecular orbitals of the two dyes were calculated using density functional theory (DFT) at the B3LYP/6-31G(d, p) level. The DFT calculations indicate that for the two dyes the HOMOs were mostly localized on the diphenylamine-carbazole moiety containing a thiophene ring (Fig. 3, see Fig. S3 in ESI† for OUK-1). On the other hand, the LUMOs were mostly localized on the thienylpyrazine moiety. Accordingly, the DFT calculations reveal that dye excitations upon light irradiation induce a strong ICT from the diphenylamine-carbazole moiety to the pyrazine moiety.
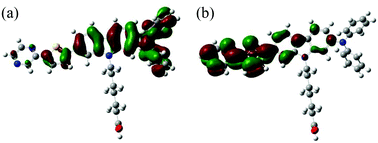 |
| | Fig. 3 (a) HOMO and (b) LUMO of OUK-3 by the density functional theory (DFT) calculations at the B3LYP/6-31G(d, p) level. | |
FTIR spectra
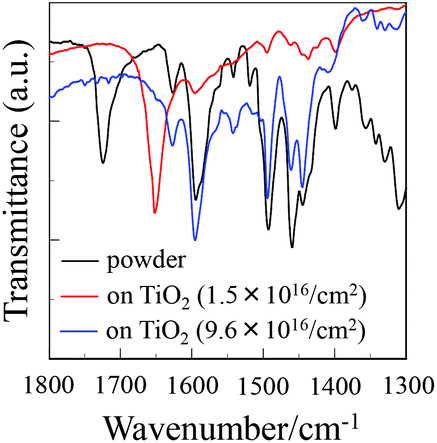
To elucidate the adsorption states of OUK-1 and OUK-3 on TiO2 nanoparticles, we measured the FTIR spectra of the dye powders and the dyes adsorbed on TiO2 nanoparticles (Fig. 4, see Fig. S4 in ESI† for OUK-1). For the dye powders of OUK-3, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band of the carboxyl group was observed at 1724 cm−1. When the dye OUK-3 was adsorbed on the TiO2 surface, the CO stretching bands of the carboxyl group disappeared completely; this indicates the formation of a bidentate bridging linkage between the carboxyl group of the dye and the Brønsted acid site on the TiO2 surface. In addition, for the powders of the two dyes the CN stretching band of the pyrazine ring was clearly observed at around 1490 cm−1. Interestingly, when the two dyes were adsorbed on the TiO2 surface, where the adsorption amount of the dye adsorbed on the TiO2 electrode is low (0.8 × 1016 and 1.5 × 1016 molecules per cm2 for OUK-1 and OUK-3 respectively), the band at 1490 cm−1 disappeared completely and a new band appeared at around 1650 cm−1, which indicates the formation of a pyrazinium ion with Brønsted acid sites on the TiO2 surface.19,20 More interestingly, when the adsorption amount of the dye adsorbed on the TiO2 electrode is high (3.0 × 1016 and 9.6 × 1016 molecules per cm2 for OUK-1 and OUK-3 respectively), the CN stretching band at 1490 cm−1 is shifted by 6 and 1 cm−1 for OUK-1 and OUK-3, respectively, to a higher wavenumber, that is, the band can be assigned to the hydrogen-bonded pyrazyl group to Brønsted acid sites on the TiO2 surface. These observations indicate that in the initial stage of dye adsorption both the dyes OUK-1 and OUK-3 are predominantly adsorbed on the TiO2 surface through the formation of a pyrazinium ion with Brønsted acid sites. However, both the dyes are adsorbed on the TiO2 surface through the hydrogen bonding at Brønsted acid sites as the dye adsorption progresses, although there still remain dye molecules adsorbed on the TiO2 surface through the formation of a pyrazinium ion with Brønsted acid sites. Consequently, the dye OUK-1 is adsorbed on the TiO2 surface through formations of both hydrogen bonding of the pyrazyl group and the pyrazinium ion at Brønsted acid sites on the TiO2 surface. On the other hand, the dye OUK-3 is adsorbed on the TiO2 surface through not only the bidentate bridging linkage of the carboxyl group but also the formations of hydrogen bonding of the pyrazyl group and the pyrazinium ion at Brønsted acid sites on the TiO2 surface. Thus, these results indicate that effective surface coverage of the TiO2 electrode is achieved successfully by employing the functionally separated D–π-A dyes with two functional groups (cyclic azine and carboxyl group) possessing bonding ability to two points on Brønsted acid sites on the TiO2 surface. Moreover, it is worth mentioning here that the functionally separated D–π-A dye sensitizers with a pyridyl group and a carboxyl group as an additional anchoring group was adsorbed on the TiO2 surface through not only the formation of a bidentate bridging linkage between the carboxyl group of the dye and the Brønsted acid site on the TiO2 surface, but also the coordinate bonding between the pyridyl group of the dye and the Lewis acid site on the TiO2 surface.12c Our work suggest that the binding modes of D–π-A dye sensitizers with cyclic azine on the TiO2 surface can be changed by controlling the basicity and the electron density of cyclic azine.
O stretching band of the carboxyl group was observed at 1724 cm−1. When the dye OUK-3 was adsorbed on the TiO2 surface, the CO stretching bands of the carboxyl group disappeared completely; this indicates the formation of a bidentate bridging linkage between the carboxyl group of the dye and the Brønsted acid site on the TiO2 surface. In addition, for the powders of the two dyes the CN stretching band of the pyrazine ring was clearly observed at around 1490 cm−1. Interestingly, when the two dyes were adsorbed on the TiO2 surface, where the adsorption amount of the dye adsorbed on the TiO2 electrode is low (0.8 × 1016 and 1.5 × 1016 molecules per cm2 for OUK-1 and OUK-3 respectively), the band at 1490 cm−1 disappeared completely and a new band appeared at around 1650 cm−1, which indicates the formation of a pyrazinium ion with Brønsted acid sites on the TiO2 surface.19,20 More interestingly, when the adsorption amount of the dye adsorbed on the TiO2 electrode is high (3.0 × 1016 and 9.6 × 1016 molecules per cm2 for OUK-1 and OUK-3 respectively), the CN stretching band at 1490 cm−1 is shifted by 6 and 1 cm−1 for OUK-1 and OUK-3, respectively, to a higher wavenumber, that is, the band can be assigned to the hydrogen-bonded pyrazyl group to Brønsted acid sites on the TiO2 surface. These observations indicate that in the initial stage of dye adsorption both the dyes OUK-1 and OUK-3 are predominantly adsorbed on the TiO2 surface through the formation of a pyrazinium ion with Brønsted acid sites. However, both the dyes are adsorbed on the TiO2 surface through the hydrogen bonding at Brønsted acid sites as the dye adsorption progresses, although there still remain dye molecules adsorbed on the TiO2 surface through the formation of a pyrazinium ion with Brønsted acid sites. Consequently, the dye OUK-1 is adsorbed on the TiO2 surface through formations of both hydrogen bonding of the pyrazyl group and the pyrazinium ion at Brønsted acid sites on the TiO2 surface. On the other hand, the dye OUK-3 is adsorbed on the TiO2 surface through not only the bidentate bridging linkage of the carboxyl group but also the formations of hydrogen bonding of the pyrazyl group and the pyrazinium ion at Brønsted acid sites on the TiO2 surface. Thus, these results indicate that effective surface coverage of the TiO2 electrode is achieved successfully by employing the functionally separated D–π-A dyes with two functional groups (cyclic azine and carboxyl group) possessing bonding ability to two points on Brønsted acid sites on the TiO2 surface. Moreover, it is worth mentioning here that the functionally separated D–π-A dye sensitizers with a pyridyl group and a carboxyl group as an additional anchoring group was adsorbed on the TiO2 surface through not only the formation of a bidentate bridging linkage between the carboxyl group of the dye and the Brønsted acid site on the TiO2 surface, but also the coordinate bonding between the pyridyl group of the dye and the Lewis acid site on the TiO2 surface.12c Our work suggest that the binding modes of D–π-A dye sensitizers with cyclic azine on the TiO2 surface can be changed by controlling the basicity and the electron density of cyclic azine.
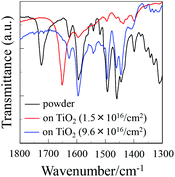 |
| | Fig. 4 FTIR spectra of the dye powders and the dye (1.5 × 1016 and 9.6 × 1016 molecules per cm2) adsorbed on TiO2 nanoparticles for OUK-3. | |
Dye-sensitized solar cells
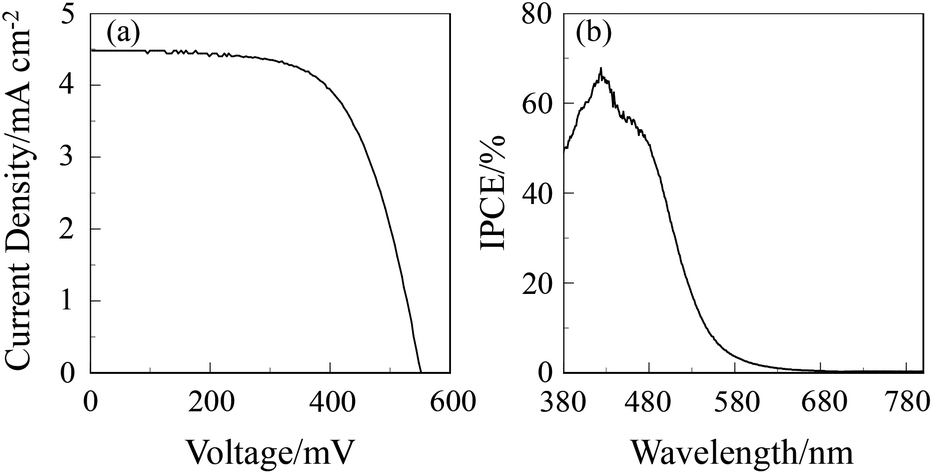
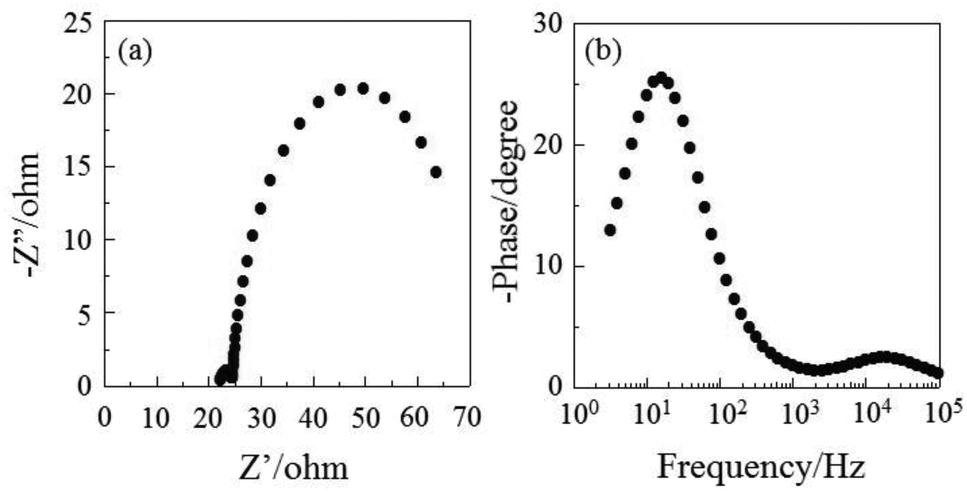
The DSSC was prepared using the dye-adsorbed TiO2 electrode (9 μm), Pt-coated glass as a counter electrode, and an acetonitrile solution with iodine (0.05 M), lithium iodide (0.1 M), and 1,2-dimethyl-3-propylimidazolium iodide (0.6 M) as an electrolyte. The photocurrent–voltage (I–V) characteristics were measured under simulated solar light (AM 1.5, 100 mW cm–2). The I–V curve and the incident photon-to-current conversion efficiency (IPCE) spectrum of DSSC based on the dye OUK-3 are shown in Fig. 5 (see Fig. S5 in ESI† for OUK-1). The photovoltaic performance parameters are collected in Table 1. The maximum adsorption amount of dyes adsorbed on TiO2 for OUK-3 is 3 times as much as that of OUK-1 (3.0 × 1017 and 9.6 × 1016 molecules per cm2 for OUK-1 and OUK-3 respectively). The high adsorption ability of OUK-3 relative to OUK-1 is attributed to the formation of a bidentate bridging linkage between the carboxyl group of OUK-3 and Brønsted acid sites on the TiO2 surface. The I–V curves show that the short-circuit photocurrent density (Jsc) and solar energy-to-electricity conversion yield (η) of OUK-3 (4.48 mA cm−2 and 1.58%) are higher than that of OUK-1 (2.99 mA cm−2 and 0.89%) (Fig. 5a). The maximum IPCE value of OUK-3 is 65% at 420 nm, which is higher than that (38% at 420 nm) of OUK-1 (Fig. 5b). Moreover, it is worth mentioning here that the Voc value of OUK-3 (550 mV) is higher than that of OUK-1 (448 mV). Thus, electrochemical impedance spectroscopy (EIS) analysis was performed to study the electron recombination process in DSSCs based on OUK-1 and OUK-3 in the dark under a forward bias of −0.60 V with a frequency range of 10 mHz to 100 kHz. The large semicircle in the Nyquist plot (Fig. 6a, see Fig. S6a in ESI† for OUK-1), which corresponds to the midfrequency peaks in the Bode phase plots, represents the charge recombination between the injected electrons in TiO2 and I3− ions in the electrolyte, that is, the charge-transfer resistances at the TiO2/dye/electrolyte interface. The Nyquist plots show that the resistance value for the large semicircle for OUK-3 (50 Ω) is larger than that of OUK-1 (26 Ω), indicating that the electron recombination resistance of OUK-3 is higher than that of OUK-1. The electron recombination lifetimes (τe) expressing the electron recombination between the injected electrons in TiO2 and I3− ions in the electrolyte, extracted from the angular frequency (ωrec) at the midfrequency peak in the Bode phase plot (Fig. 6b, see Fig. S6b in ESI† for OUK-1) using τe = 1/ωrec, are 11 ms for DSSC based on OUK-3, which is slightly larger than 10 ms for DSSC based on OUK-1. This result revealed that the charge recombination between the injected electrons in TiO2 and I3− ions in the electrolyte is not the major reason for the difference in the Voc value between the two dyes, but the increase in the number of injected electrons in the CB of TiO2 and/or a negative shift of the Ecb of TiO2 by the formation of hydrogen bonding of the pyrazyl group at Brønsted acid sites on the TiO2 surface with the increasing dye loading of OUK-3 on the TiO2 electrode may result in a higher Voc value for OUK-3.4,6 Consequently, the higher photovoltaic performance of OUK-3 is attributed to the improvement of light-harvesting efficiency (LHE) and the increase in the number of injected electrons in the CB of TiO2 due to a high adsorption ability of the functionally separated D–π-A dye onto the TiO2 electrode.
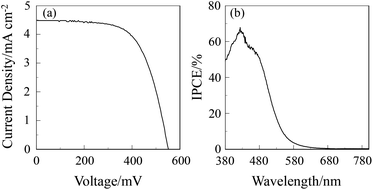 |
| | Fig. 5 (a) I–V curve and (b) IPCE spectrum of DSSC based on OUK-3. | |
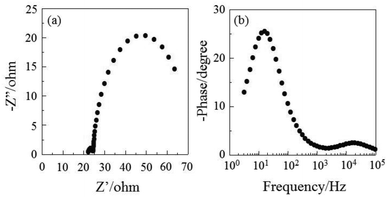 |
| | Fig. 6 (a) Nyquist plot and (b) Bode phase plot of DSSCs based on OUK-3. | |
Conclusions
We have designed and synthesized a functionally separated D–π-A dye sensitizer OUK-3 with a pyrazyl group as an electron-withdrawing anchoring group and a carboxyl group as an additional anchoring group for dye-sensitized solar cells. The dye OUK-3 is adsorbed on the TiO2 surface through not only the bidentate bridging linkage of the carboxyl group but also the formation of hydrogen bonding of the pyrazyl group and the pyrazinium ion at Brønsted acid sites on the TiO2 surface. This work revealed that functionally separated D–π-A dye sensitizers can achieve effective surface coverage of the TiO2 electrode due to its high adsorption ability onto the TiO2 electrode, leading to not only the improvement of light-harvesting efficiency, but also the increase in the number of injected electrons in the CB of TiO2, which is responsible for the higher Voc value of functionally separated D–π-A dye sensitizers. This work also shows that the functionally separated D–π-A dyes with two functional groups (cyclic azine and carboxyl group) possessing bonding ability to two points on the acid sites (Brønsted acid site and Lewis acid site) on the TiO2 surface would be expected to be one of the most promising classes of organic dye sensitizers for dye-sensitized solar cells. However, to further improve the photovoltaic performances of dye-sensitized solar cells based on the functionally separated D–π-A dye sensitizers, the extension of the absorption spectrum of functionally separated D–π-A dye sensitizers is necessary; for example, the destabilization of the HOMO level by the introduction of a stronger electron-donating group such as starburst triarylamine and the expansion of the π-conjugated system by the introduction of a long π-bridge such as terthiophene is one of the most effective strategies to improve the light-harvesting efficiency.
Experimental
General
Melting points were measured with a Yanaco micro melting point apparatus MP model. TG-DTA was carried out on a SII EXTRA6000 thermal analyser. IR spectra were recorded on a Perkin Elmer Spectrum One FT-IR spectrometer by the ATR method. High-resolution mass spectral data were acquired on a Thermo Fisher Scientific LTQ Orbitrap XL. 1H NMR spectra were recorded on a Varian-400 (400 MHz) FT NMR spectrometer with tetramethylsilane as an internal standard. Absorption spectra were observed with a Shimadzu UV-3150 spectrophotometer and fluorescence spectra were measured with a HORIBA FluoroMax-4 spectrofluorometer. The fluorescence quantum yields in solution were determined by a HORIBA FluoroMax-4 spectrofluorometer by using a calibrated integrating sphere system (λex = 402 nm for OUK-1 and OUK-3). Cyclic voltammetry (CV) curves were recorded in the DMF/Bu4NClO4 (0.1 M) solution with a three-electrode system consisting of Ag/Ag+ as the reference electrode, Pt plate as a working electrode, and Pt wire as a counter electrode by using a AMETEK Versa STAT 4 potentiostat. The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels of OUK-1 and OUK-3 were evaluated from the spectral analyses and the CV data. The HOMO energy level was evaluated from the Eox1/2. The LUMO energy level was estimated from the Eox1/2 and an intersection of absorption and fluorescence spectra (445 nm; 2.79 eV for OUK-1 and 443 nm; 2.80 eV for OUK-3), which correspond to the energy gap between the HOMO and the LUMO. Electrochemical impedance spectroscopy (EIS) for DSSCs in the dark under a forward bias of −0.60 V with a frequency range of 10 mHz to 100 kHz was measured with a AMETEK Versa STAT 3.
Synthesis
N,N-Diphenyl-7-(5-(trimethylstannyl)thiophen-2-yl)-9H-carbazol-2-amine (2). To a THF solution (1.2 mL) of 119 (0.1 g, 0.24 mmol) under an argon atmosphere was added 1.6 M hexane solution of nBuLi (0.3 ml) at −65 °C. After stirring for 1 h, Me3SnCl (0.075 g, 0.38 mmol) was added and the solution was stirred at −65 °C for 2 h. After that, the solution was stirred for 8 h at room temperature. The reaction was quenched with water, and then the solution was extracted with dichloromethane. The dichloromethane extract was evaporated under reduced pressure. The resulting residue was subjected to reprecipitation from dichloromethane–hexane to give 2 (0.11 g, yield 78%) as a green powder; IR (ATR): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 3417, 1583, 1486 cm−1; 1H NMR (400 MHz, acetone-d6, TMS) δ = 0.40 (s, 9H), 6.93 (dd, J = 2.0 and 8.4 Hz, 1H), 7.02–7.05 (m, 2H), 7.09–7.11 (m, 4H), 7.15 (d, J = 2.0 Hz, 1H), 7.24 (d, J = 3.4 Hz, 1H), 7.28–7.32 (m, 4H), 7.64 (dd, J = 1.6 and 8.2 Hz, 1H), 7.57 (d, J = 3.4 Hz, 1H), 7.75 (d, J = 1.0 Hz, 1H), 8.01 (d, J = 8.4 Hz, 1H), 8.05 (d, J = 8.2 Hz, 1H), 10.21 (s, –NH); HRMS (ESI): m/z (%):[M+˙] calcd for C31H28N2SSn, 580.09897; found 580.09906.
= 3417, 1583, 1486 cm−1; 1H NMR (400 MHz, acetone-d6, TMS) δ = 0.40 (s, 9H), 6.93 (dd, J = 2.0 and 8.4 Hz, 1H), 7.02–7.05 (m, 2H), 7.09–7.11 (m, 4H), 7.15 (d, J = 2.0 Hz, 1H), 7.24 (d, J = 3.4 Hz, 1H), 7.28–7.32 (m, 4H), 7.64 (dd, J = 1.6 and 8.2 Hz, 1H), 7.57 (d, J = 3.4 Hz, 1H), 7.75 (d, J = 1.0 Hz, 1H), 8.01 (d, J = 8.4 Hz, 1H), 8.05 (d, J = 8.2 Hz, 1H), 10.21 (s, –NH); HRMS (ESI): m/z (%):[M+˙] calcd for C31H28N2SSn, 580.09897; found 580.09906.
N,N-Diphenyl-7-(5-(pyrazin-2-yl)thiophen-2-yl)-9H-carbazol-2-amine (3). A solution of 2 (0.1 g, 0.18 mmol), 2-iodopyrazine 0.028 ml, 0.28 mmol), and Pd(PPh3)4 (0.014 g, 0.001 mmol) in toluene (2 mL) was stirred for 10 h at 110 °C under an argon atmosphere. The reaction mixture was diluted with water, and then the solution was extracted with dichloromethane. The dichloromethane extract was evaporated under reduced pressure. The residue was chromatographed on silica gel (dichloromethane as the eluent) to give 3 (0.07 g, yield 79%) as a yellow solid; m.p. 298–299 °C; IR (ATR): = 3168, 1584, 1489 cm−1; 1H NMR (400 MHz, acetone-d6, TMS) δ = 6.96 (dd, J = 2.0 and 8.4 Hz, 1H), 7.03–7.08 (m, 2H), 7.11–7.13 (m, 4H), 7.17 (d, J = 2.0 Hz, 1H), 7.29–7.34 (m, 4H), 7.60–7.63 (m, 2H), 7.86 (d, J = 1.2 Hz, 1H), 7.95 (d, J = 4.0 Hz, 1H), 8.04 (d, J = 8.8 Hz, 1H), 8.12 (d, J = 8.0 Hz, 1H), 8.47 (d, J = 2.4 Hz, 1H), 8.56 (dd, J = 1.6 and 2.4 Hz, 1H), 9.18 (d, J = 1.6 Hz, 1H), 10.31 (s, –NH); HRMS (APCI): m/z (%):[M + H+] calcd for C32H23N4S, 495.16379; found 495.16351.
Ethyl 7-(2-(diphenylamino)-7-(5-(pyrazin-2-yl)thiophen-2-yl)-9H-carbazol-9-yl)heptanoate (4). A solution of 3 (0.057 g, 0.12 mmol) in DMF (1.5 ml) was treated with sodium hydride (60%, 0.014 g, 0.58 mmol) and stirred for 1 h at room temperature. Ethyl 7-bromoheptanoate (0.0067 ml, 0.35 mmol) was added dropwise and the solution was stirred at room temperature for 3 h. The reaction was quenched with water, and then the solution was extracted with dichloromethane. The dichloromethane extract was evaporated under reduced pressure. The residue was chromatographed on silica gel (dichloromethane as the eluent) to give 4 (0.06 g, yield 79%) as a yellow viscous solid; IR (ATR): = 1726, 1593, 1459 cm−1; 1H NMR (400 MHz, acetone-d6, TMS) δ = 1.17 (t, J = 7.2 Hz, 3H), 1.31–1.34 (m, 4H), 1.49–1.54 (m, 2H), 1.82–1.86 (m, 2H), 2.21 (t, J = 7.2 Hz, 2H), 4.01–4.06 (q, 2H), 4.39 (t, J = 6.8 Hz, 2H), 6.94 (dd, J = 1.8 and 8.5 Hz, 1H), 7.03–7.07 (m, 2H), 7.11–7.14 (m, 4H), 7.25 (d, J = 1.8 Hz, 1H), 7.29–7.34 (m, 4H), 7.62 (dd, J = 1.6 and 8.1 Hz, 1H), 7.67 (d, J = 3.9 Hz, 1H), 7.92 (d, J = 1.1 Hz, 1H), 7.95 (d, J = 3.9 Hz, 1H), 8.05 (d, J = 8.5 Hz, 1H), 8.13 (d, J = 8.1 Hz, 1H), 8.47 (d, J = 2.5 Hz, 1H), 8.56 (dd, J = 1.6 and 2.5 Hz, 1H), 9.18 (d, J = 1.6 Hz, 1H); HRMS (APCI): m/z (%):[M + H+] calcd for C41H39O2N4S, 651.27882; found 651.27863.
7-(2-(Diphenylamino)-7-(5-(pyrazin-2-yl)thiophen-2-yl)-9H-carbazol-9-yl)heptanoic acid (OUK-3). To a solution of 4 (0.068 g, 0.1 mmol) in the mixed solvent of ethanol (27 ml) and dichloromethane (10 ml) was added dropwise aqueous NaOH (10 ml, 0.05 M) with stirring at 75 °C. After further stirring for 17 h, the solution was concentrated under reduced pressure. The residue was dissolved in dichloromethane, and washed with water, in which the aqueous layer was acidified to pH 3 with 2 M HCl. The dichloromethane extract was evaporated under reduced pressure. The resulting residue was subjected to reprecipitation from dichloromethane–hexane to give OUK-3 (0.05 g, yield 77%) as a yellow powder; m.p. 129–131 °C, d.p. 376 °C; IR (ATR): = 1724, 1594, 1459 cm−1; 1H NMR (400 MHz, acetone-d6, TMS) δ = 1.32–1.35 (m, 4H), 1.50–1.54 (m, 2H), 1.82–1.86 (m, 2H), 2.22 (t, J = 7.4 Hz, 2H), 4.40 (t, J = 6.8 Hz, 2H), 6.94 (dd, J = 1.7 and 8.4 Hz, 1H), 7.03–7.07 (m, 2H), 7.11–7.14 (m, 4H), 7.25 (s, 1H), 7.29–7.34 (m, 4H), 7.61 (d, J = 6.8 Hz, 1H), 7.67 (d, J = 3.9 Hz, 1H), 7.93–7.95 (m, 2H), 8.05 (d, J = 8.4 Hz, 1H), 8.12 (d, J = 8.2 Hz, 1H), 8.46 (s, 1H), 8.56 (s, 1H), 9.18 (s, 1H); HRMS (APCI): m/z (%):[M + H+] calcd for C39H35O2N4S, 623.24752; found 623.24744.
Preparation of DSSCs based on dyes OUK-1 and OUK-3
The TiO2 paste (JGC Catalysts and Chemicals Ltd, PST-18NR) was deposited on a fluorine-doped-tin-oxide (FTO) substrate by doctor-blading, and sintered for 50 min at 450 °C. The 9 μm thick TiO2 electrode was immersed into a 1 mM dye solution in THF for 15 hours sufficient to adsorb the photosensitizer. The DSSCs were fabricated by using the TiO2 electrode (0.5 × 0.5 cm2 in the photoactive area) thus prepared, with Pt-coated glass as a counter electrode, and a solution of 0.05 M iodine, 0.1 M lithium iodide, and 0.6 M 1,2-dimethyl-3-propylimidazolium iodide in acetonitrile as the electrolyte. The photocurrent–voltage characteristics were measured using a potentiostat under a simulated solar light (AM 1.5, 100 mW cm−2). IPCE spectra were measured under monochromatic irradiation with a tungsten-halogen lamp and a monochromator. The dye-coated film was immersed in a mixed solvent of THF–DMSO–NaOH aq. 1 M (5:4:1), which was used to determine the amount of dye molecules adsorbed onto the film by measuring the absorbance. The quantification of the dye was made based on the λmax (405 nm for OUK-1 and 402 nm for OUK-3) and the molar extinction coefficient of the dye in the above solution. Absorption spectra of the dyes adsorbed on TiO2 nanoparticles were recorded on the dye-adsorbed TiO2 film in the transmission mode with a calibrated integrating sphere system.
Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science (JSPS) KAKENHI grant number 24550225 and by Hodogaya Chemical Co., Ltd.
Notes and references
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed.
-
(a) T. Bessho, S. M. Zakeeruddin, C.-Y. Yeh, E. W.-G. Diau and M. Grätzel, Angew. Chem., Int. Ed., 2010, 49, 6646 CrossRef CAS PubMed;
(b) A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629 CrossRef CAS PubMed;
(c) L.-L. Li and E. W.-G. Diau, Chem. Soc. Rev., 2013, 42, 291 RSC;
(d) S. Mathew, A. Yella, P. Gao, R. Humphry-Baker, B. F. E. Curchod, N. Ashari-Astani, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, Nat. Chem., 2014, 6, 242 CrossRef CAS PubMed;
(e) A. Yella, C.-L. Mai, S. M. Zakeeruddin, S.-N. Chang, C.-H. Hsieh, C.-Y. Yeh and M. Grätzel, Angew. Chem., Int. Ed., 2014, 53, 2973 CrossRef CAS PubMed.
-
(a) Z. Ning and H. Tian, Chem. Commun., 2009, 5483 RSC;
(b) Z. Ning, Y. Fu and H. Tian, Energy Environ. Sci., 2010, 3, 1170 RSC.
- A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474 CrossRef CAS PubMed.
-
(a) Y. Ooyama and Y. Harima, Eur. J. Org. Chem., 2009, 18, 2903 CrossRef;
(b) Y. Ooyama and Y. Harima, ChemPhysChem, 2012, 13, 4032 CrossRef CAS PubMed.
- K. Ladomenou, T. N. Kitsopoulos, G. D. Sharma and A. G. Coutsolelos, RSC Adv., 2014, 4, 21379 RSC.
- N. Manfredi, B. Cecconi and A. Abbotto, Eur. J. Org. Chem., 2014, 7069 CrossRef CAS.
-
(a) X. Wang, J. Yang, H. Yu, F. Li, L. Fan, W. Sun, Y. Liu, Z. Y. Koh, J. Pan, W.-L. Yim, L. yan and Q. Wang, Chem. Commun., 2014, 50, 3965 RSC;
(b) S.-G. Li, K.-J. Jiang, J.-H. Huang, L.-M. Yang and Y.-L. Song, Chem. Commun., 2014, 50, 4309 RSC;
(c) D. K. Panda, F. S. Goodson, S. Ray and S. Saha, Chem. Commun., 2014, 50, 5358 RSC;
(d) K. Kakiage, Y. Aoyama, T. Yano, T. Otsuka, T. Kyomen, M. Unno and M. Hanaya, Chem. Commun., 2014, 50, 6379 RSC;
(e) A. Amacher, C. Yi, J. yang, M. P. Bircher, Y. Fu, M. Cascella, M. Grätzel, S. Decurtins and S.-X. Liu, Chem. Commun., 2014, 50, 6540 RSC;
(f) J. Yang, P. Ganesan, J. Teuscher, T. Moehl, Y. J. Kim, C. Yi, P. Comte, K. Pei, T. W. Holcombe, M. K. Nazeeruddin, J. Hua, S. K. Zakeeruddin, H. Tian and M. Grätzel, J. Am. Chem. Soc., 2014, 136, 5772 Search PubMed.
-
(a) R. B. Ambre, G.-F. Chang and C.-H. Hung, Chem. Commun., 2014, 50, 725 RSC;
(b) T. Ikeuchi, H. Nomoto, N. Masaki, M. J. Griffith, S. Mori and M. Kimura, Chem. Commun., 2014, 50, 1941 RSC;
(c) R. Agosta, R. Grisorio, L. De Marco, G. Romanazzi, G. P. Suranna, G. Gigli and M. Manca, Chem. Commun., 2014, 50, 9451 RSC;
(d) A. Dessi, M. Calamante, A. Mordini, M. Peruzzin, A. Sinicropi, R. Basosi, F. F. de Biani, M. Taddei, D. Colonna, A. D. Carlo, G. Reginato and L. Zani, Chem. Commun., 2014, 50, 13952 RSC;
(e) X. Sun, Y. Wang, X. Li, H. Ågren, W. Zhu, H. Tian and Y. Xie, Chem. Commun., 2014, 50, 15609 RSC.
- J. Mao, N. He, Z. Ning, Q. Zhang, F. Guo, L. Chen, W. Wu, J. Hua and H. Tian, Angew. Chem., Int. Ed., 2012, 51, 9873 CrossRef CAS PubMed.
-
(a) Y. Ooyama, S. Inoue, R. Asada, G. Ito, K. Kushimoto, K. Komaguchi, I. Imae and Y. Harima, Eur. J. Org. Chem., 2010, 92 CrossRef CAS;
(b) Y. Ooyama, S. Inoue, T. Nagano, K. Kushimoto, J. Ohshita, I. Imae, K. Komaguchi and Y. Harima, Angew. Chem., Int. Ed., 2011, 50, 7429 CrossRef CAS PubMed;
(c) Y. Ooyama, T. Nagano, S. Inoue, I. Imae, K. Komaguchi, J. Ohshita and Y. Harima, Chem. – Eur. J., 2011, 17, 14837 CrossRef CAS PubMed;
(d) Y. Ooyama, N. Yamaguchi, I. Imae, K. Komaguchi, J. Ohshita and Y. Harima, Chem. Commun., 2013, 49, 2548 RSC;
(e) Y. Ooyama, Y. Hagiwara, T. Mizumo, Y. Harima and J. Ohshita, New J. Chem., 2013, 37, 2479 RSC;
(f) Y. Ooyama, T. Sato, Y. Harima and J. Ohshita, J. Mater. Chem. A, 2014, 2, 3293 RSC.
-
(a) D. Daphnomili, G. D. Sharma, S. Biswas, T. K. R. Justin and A. G. Goutsolelos, J. Photochem. Photobiol., A, 2013, 253, 88 CrossRef CAS PubMed;
(b) J. Lu, X. Xu, Z. Li, k. Cao, J. Cui, Y. Zhang, Y. Shen, Y. Li, J. Zhu, S. Dai, W. Chjen, Y. Cheng and M. Wang, Chem. – Asian J., 2013, 8, 956 CrossRef CAS PubMed;
(c) M.-D. Zhang, H.-X. Xie, X.-H. Ju, L. Qin, Q.-X. Yang, H.-G. Zheng and X.-F. Zhou, Phys. Chem. Chem. Phys., 2013, 15, 634 RSC;
(d) D. Daphnomili, G. Landrou, P. Singh, A. Thomas, K. Yesudas, B. K. G. D. Sharma and A. G. Goutsolelos, RSC Adv., 2012, 2, 12899 RSC;
(e) L. Wang, X. yang, S. Li, M. Cheng and L. Sun, RSC Adv., 2014, 4, 13677 Search PubMed;
(f) T. Sakurada, Y. Arai and H. Segawa, RSC Adv., 2014, 4, 13201 RSC;
(g) J. Mao, D. Wang, S.-H. Liu, Y. Hang, Y. Xu, Q. Zhang, W. Wu, P.-T. Chou and J. Hua, Asian J. Org. Chem., 2014, 3, 153 CrossRef CAS.
- H. He, A. Gurung and L. Si, Chem. Commun., 2012, 48, 5910 RSC.
-
(a) Y. Ooyama, Y. Shimada, Y. Kagawa, I. Imae and Y. Harima, Org. Biomol. Chem., 2007, 5, 2046 RSC;
(b) Y. Ooyama, Y. Shimada, Y. Kagawa, Y. Yamada, I. Imae, K. Komaguchi and Y. Harima, Tetrahedron Lett., 2007, 48, 9167 CrossRef CAS PubMed;
(c) Y. Ooyama, Y. Shimada, S. Inoue, T. Nagano, Y. Fujikawa, K. Komaguchi, I. Imae and Y. Harima, New J. Chem., 2011, 35, 111 RSC.
- Y. Hao, X. Yang, J. Cong, H. Tian, A. Hagfeldt and L. Sun, Chem. Commun., 2009, 4031 RSC.
- J. Cong, X. Yang, J. Liu, J. Zhao, Y. Hao, Y. Wang and L. Sun, Chem. Commun., 2012, 48, 6663 RSC.
- Y. Ooyama, Y. Hagiwara, Y. Oda, T. Mizumo, Y. Harima and J. Ohshita, New J. Chem., 2013, 37, 2336 RSC.
- Y. Ooyama, K. Uenaka, Y. Harima and J. Ohshita, RSC Adv., 2014, 4, 30225 RSC.
-
(a) T. J. Dines, L. D. MacGregor and C. H. Rochester, Phys. Chem. Chem. Phys., 2001, 3, 2676 RSC;
(b) M. I. Zaki, M. A. Hasan, F. A. Al-Sagheer and L. Pasupulety, Colloids Surf., A, 2001, 190, 261 CrossRef CAS;
(c) O. Kasende and T. Zeegers-Huyskens, J. Phys. Chem., 1984, 88, 2132 CrossRef CAS;
(d) H. Takahashi, K. Mamola and E. K. Plyler, J. Mol. Spectrosc., 1966, 21, 217 CrossRef CAS;
(e) M. A. Montańez, I. L. Tocón, J. C. Otero and J. I. Marcos, J. Mol. Struct., 1999, 482–483, 201 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Details of optical and electrochemical properties, and photovoltaic performances of compounds. See DOI: 10.1039/c5qo00050e |
|
| This journal is © the Partner Organisations 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a