DOI:
10.1039/C5QO00016E
(Review Article)
Org. Chem. Front., 2015,
2, 739-752
The Mitsunobu reaction in the 21st century
Received
13th January 2015
, Accepted 12th March 2015
First published on 17th March 2015
Abstract
The Mitsunobu reaction was first described almost fifty years ago and has enjoyed immense popularity since its inception. The purpose of this review is to focus on the more recent advances and applications of Mitsunobu chemistry, particularly from the 1990s to the present day. In addition to a discussion of newer reagents that facilitate purification, we will describe more contemporary applications of this chemistry, especially as applied to the synthesis of pharmaceuticals and their precursors.
 Steven Fletcher | Dr Steven Fletcher hails from Wolsingham, Co. Durham (UK). Steven received his MNatSci (2000) in Chemistry and MA (2003) from the University of Cambridge (UK), then his PhD in Medicinal Chemistry (2004) from Imperial College London (UK). He pursued post-doctoral studies with Professor Andrew Hamilton (Yale University, USA) and Professor Patrick Gunning (University of Toronto, Canada) before joining the University of Maryland School of Pharmacy (USA) as an Assistant Professor of Chemistry (2009). Steven's medicinal chemistry research focuses on the development of inhibitors of the proto-oncoproteins c-Myc and Mcl-1, whilst his synthetic chemistry interests lie with the Mitsunobu reaction. |
Introduction
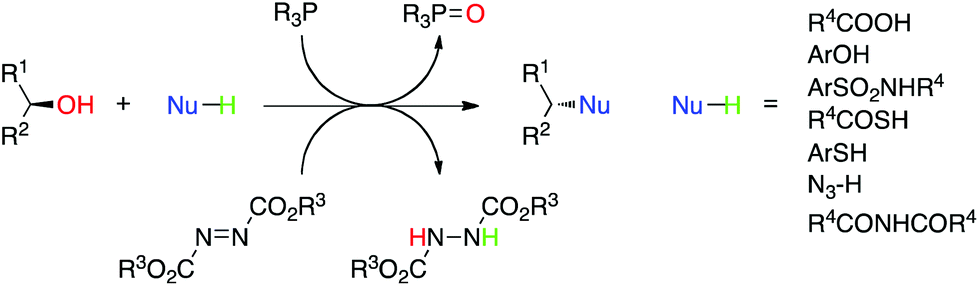
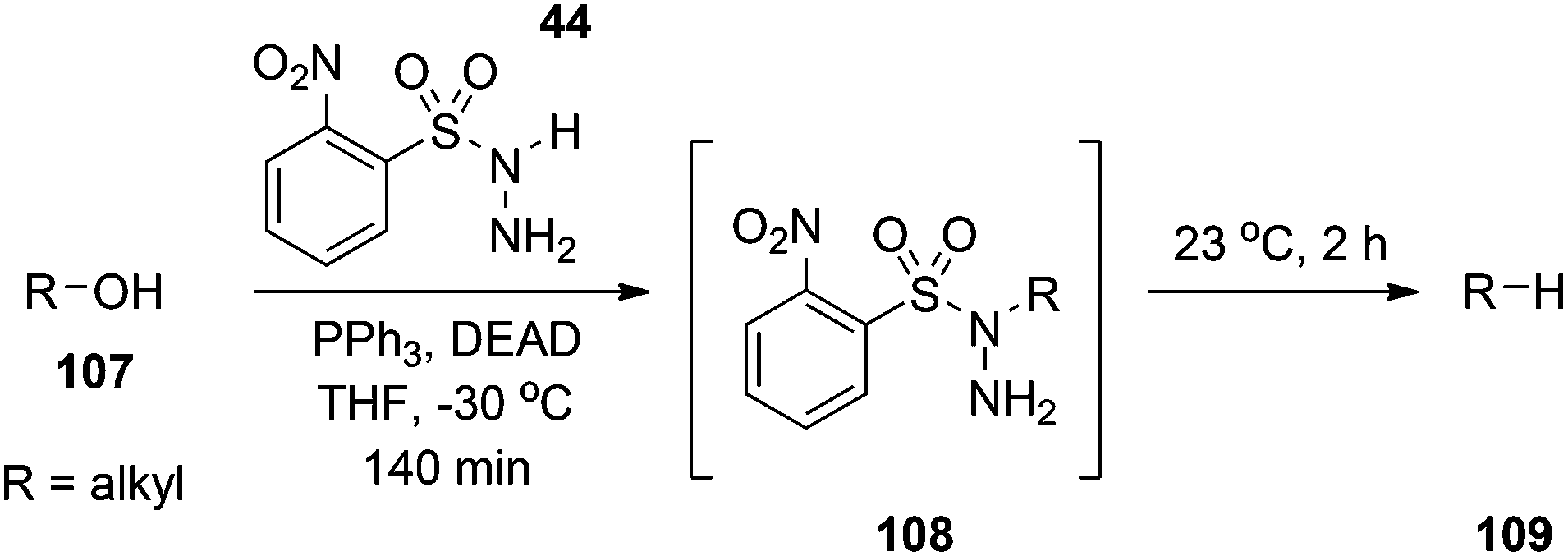
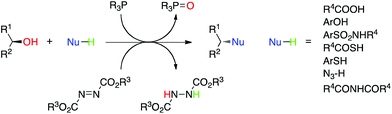
The Mitsunobu reaction is the dehydrative coupling of a primary or secondary alcohol (occasionally, tertiary alcohols have been used) to a pronucleophile (NuH), which is mediated by the reaction between a dialkyl azodicarboxylate and a trialkyl- or triarylphosphine (Scheme 1). In the process of the reaction, the azo species becomes reduced to a hydrazine derivative, whilst the phosphine is oxidized to a phosphine oxide; both by-products can plague purification strategies. The reaction is named after its discoverer Oyo Mitsunobu who first reported this chemistry in 1967.1,2 When chiral, secondary alcohols are employed, complete inversion of stereochemistry is observed in all but a few cases.3 In “conventional” Mitsunobu chemistry, there is a wide range of pronucleophiles that can participate in the reaction. Suitable pronucleophiles include (thio)carboxylic acids, (thio)phenols, imides and sulfonamides, permitting the formation of C–O, C–S and C–N bonds (and C–C bonds as will be seen shortly). It is generally agreed that the pKa of the pronucleophile must be around 11 or below for a successful reaction with the typical Mitsunobu reagents diethyl azodicarboxylate (DEAD; R3 = Et in Scheme 1) or diisopropyl azodicarboxylate (DIAD; R3 = iPr in Scheme 1) and triphenylphosphine (PPh3). This “pKa rule” stems from the mechanism of the reaction wherein the betaine that results from the reaction between DEAD and PPh3 has a pKa of about 13 and removes the acidic proton from the pronucleophile, otherwise alkylation of DEAD occurs. The Mitsunobu reaction proceeds under mild, essentially neutral conditions, and typically at 0 °C to room temperature. Standard solvents for the reaction include THF, diethyl ether, dichloromethane and toluene, although sometimes more polar solvents, including ethyl acetate, acetonitrile and DMF, have been used. The Mitsunobu reaction has been reviewed several times before.2–5 The purpose of this review is to focus on newer applications and more recent innovations associated with Mitsunobu chemistry.
 |
| | Scheme 1 The Mitsunobu reaction. | |
Contemporary Mitsunobu chemistry
Oxygen pronucleophiles
Perhaps the single, most important feature of the Mitsunobu reaction is that secondary alcohols undergo clean inversion of their stereogenic centres.1–5 Thus, hydrolysis of the ester that is yielded from the dehydrative coupling of a carboxylic acid and a secondary alcohol will furnish the inverted alcohol generally in high enantiomeric purity. In addition to carboxylic acids, other oxygen pronucleophiles that have become popular coupling partners in the Mitsunobu reaction include phenols and also alcohols themselves in intramolecular reactions. Below are some newer oxygen pronucleophiles that have been successfully introduced into this reaction.
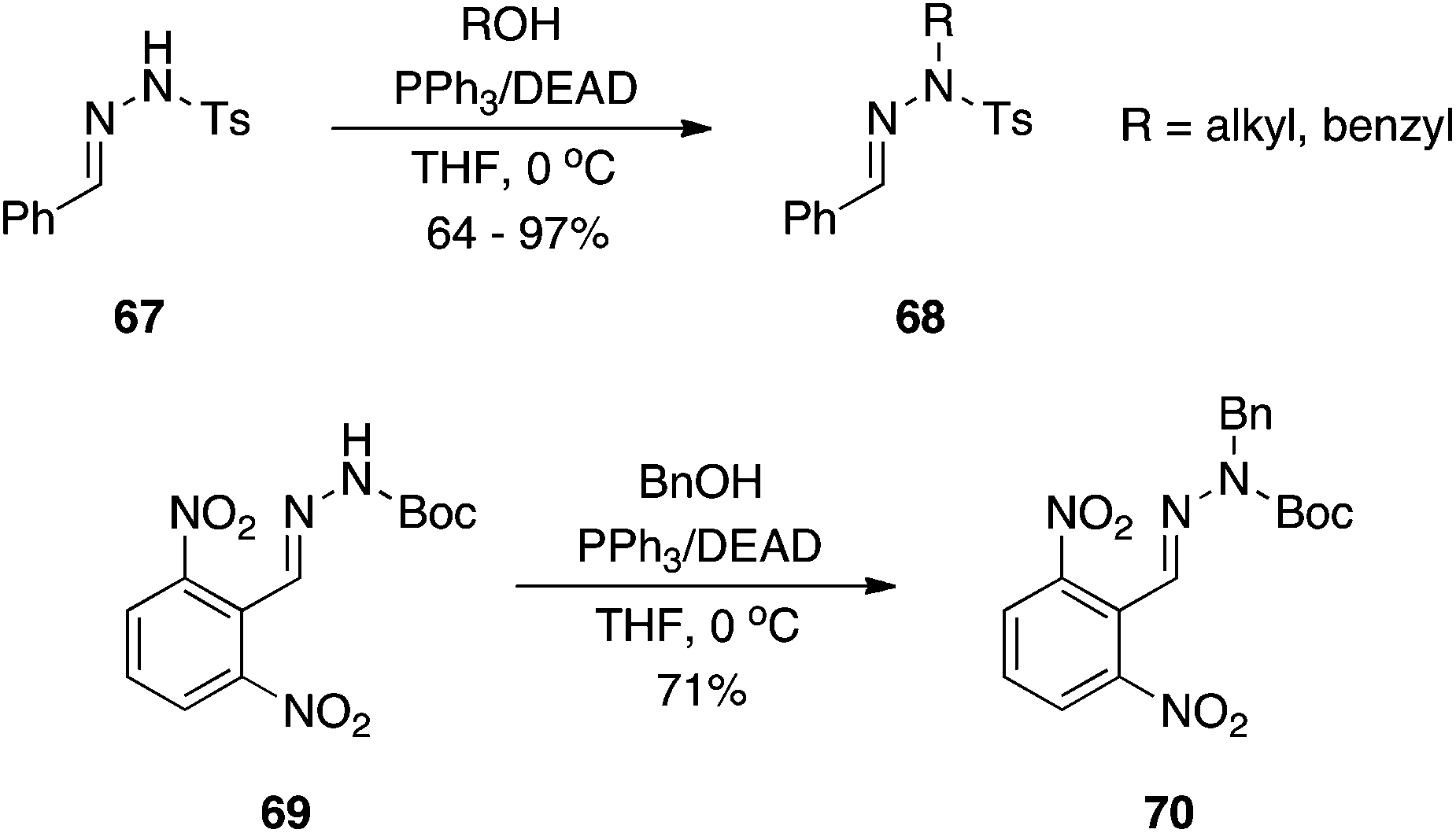
Oximes.
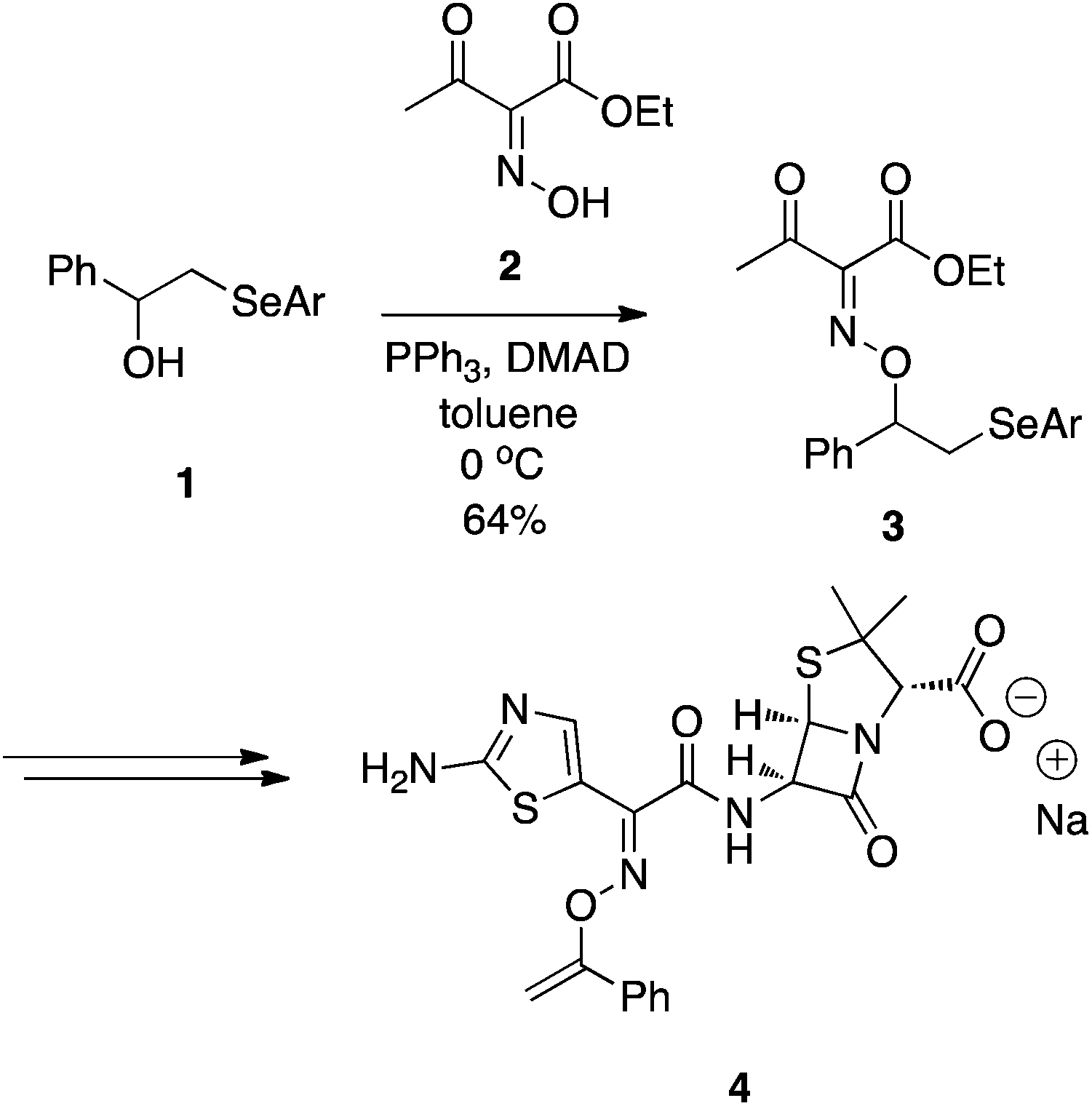
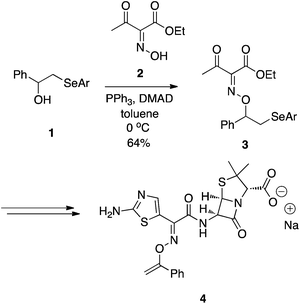
En route to some O-vinyl penicillin derivatives, such as 4, Stachulski at SmithKline Beecham demonstrated that oximes function well as oxygen pronucleophiles (Scheme 2).6 In place of the usual DEAD or DIAD, dimethyl acetylenedicarboxylate (DMAD) was used alongside PPh3. Akin to DIAD, DMAD can also undergo a Michael-type reaction with phosphines to yield an activated betaine. Although the Mitsunobu reaction proceeded in only moderate yield (64%), it is unclear if attempts were made to optimize the reaction.
 |
| | Scheme 2 Oximes as oxygen pronucleophiles in the Mitsunobu reaction. | |
2-Amidophenols: synthesis of benzoxazoles.
Motivated by the prevalence of the benzoxazole motif in a range of anti-bacterials, anti-parasitics and H2-antagonists,7 Wang and Hauske wanted to explore this pharmacophoric scaffold through combinatorial synthesis on solid phase.8 However, the traditional approaches of thermal cyclization of a 2-amidophenol derivative with acidic reagents such as SOCl2 proved incompatible with a solid support. Thus, the authors looked to the Mitsunobu reaction which they reasoned would accomplish the same goal of cyclodehydration but under mild and neutral conditions. As illustrated in Scheme 3, this new application of Mitsunobu chemistry proceeded in high yield, providing a gentler alternative to the existing methods for benzoxazole synthesis.
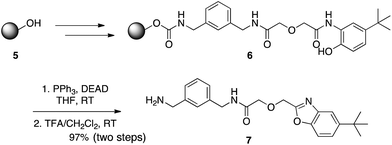 |
| | Scheme 3 2-Amidophenols undergo cyclodehydrations with DEAD–PPh3 to furnish benzoxazoles. | |
2-Ureidophenols: synthesis of N2-substituted benzoxazoles.
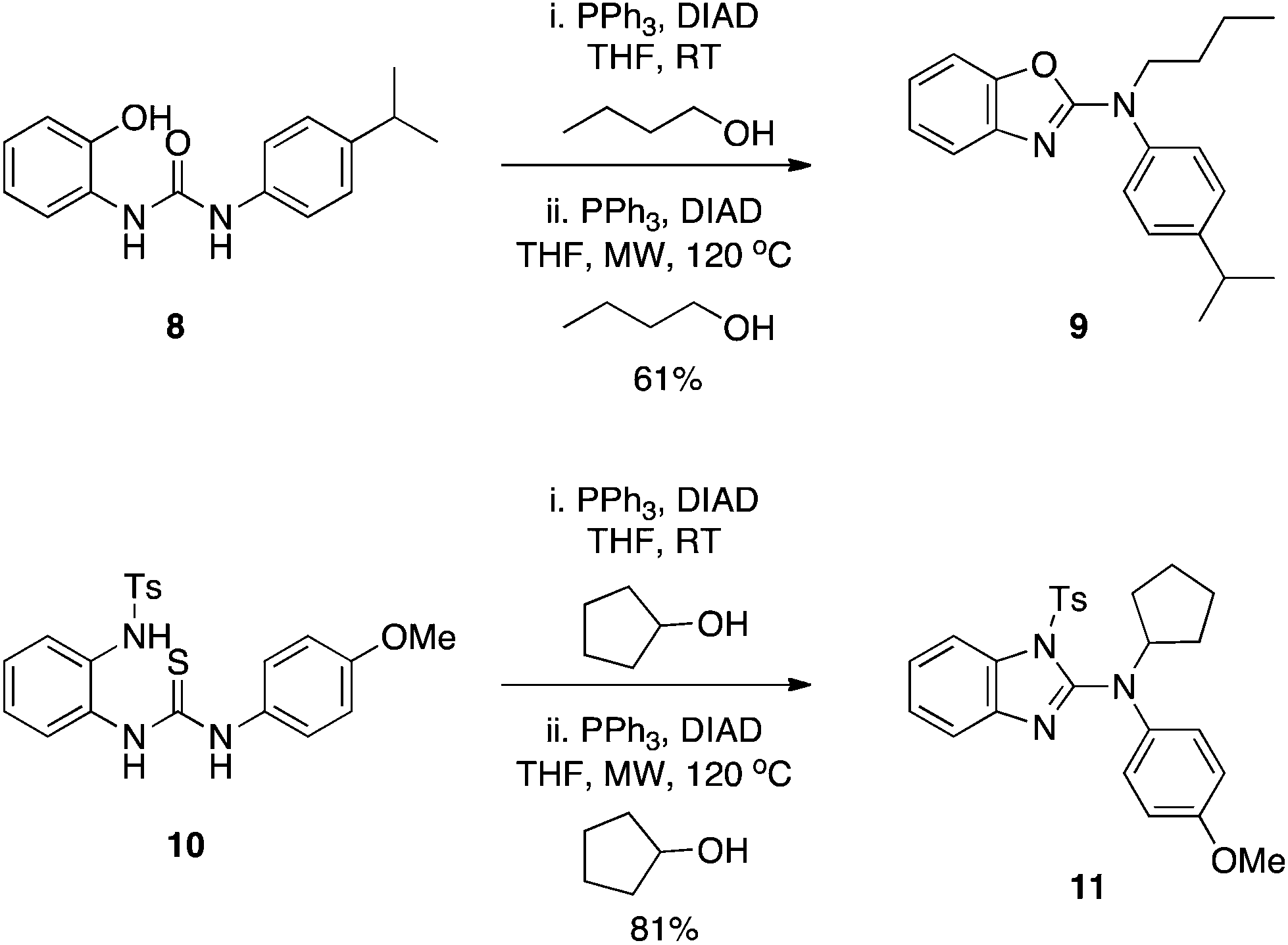
Yan and colleagues inadvertently took Wang's work further.9 Initially intending to alkylate the phenol OH of 2-ureidophenol 8, the isolated product was instead the N2-substituted benzoxazole 9 (Scheme 4) via a cascade of two Mitsunobu reactions in one pot.9 To account for the formation of 9, the authors proposed that the hydroxyl within the –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(OH)–NH– tautomeric form of the urea group played the role of the alcohol, which was turned into a good leaving group during the course of the reaction and was subsequently displaced by the phenoxide ion to generate the benzoxazole scaffold. A second Mitsunobu reaction under microwave (MW) conditions and at elevated temperature (optimized conditions) then alkylated the exocyclic nitrogen to deliver the observed product 9. The chemistry appears quite general, and was also successful with the corresponding tosyl-activated anilines (as a substitute for the phenol OH) to yield the analogous N2-substituted benzimidazoles, for example 11. It is noteworthy that, according to the proposed mechanism, the reaction to generate 11 sees a thiol (of the appropriate tautomer of the thiourea group) functioning as the alcohol in the Mitsunobu reaction.
C(OH)–NH– tautomeric form of the urea group played the role of the alcohol, which was turned into a good leaving group during the course of the reaction and was subsequently displaced by the phenoxide ion to generate the benzoxazole scaffold. A second Mitsunobu reaction under microwave (MW) conditions and at elevated temperature (optimized conditions) then alkylated the exocyclic nitrogen to deliver the observed product 9. The chemistry appears quite general, and was also successful with the corresponding tosyl-activated anilines (as a substitute for the phenol OH) to yield the analogous N2-substituted benzimidazoles, for example 11. It is noteworthy that, according to the proposed mechanism, the reaction to generate 11 sees a thiol (of the appropriate tautomer of the thiourea group) functioning as the alcohol in the Mitsunobu reaction.
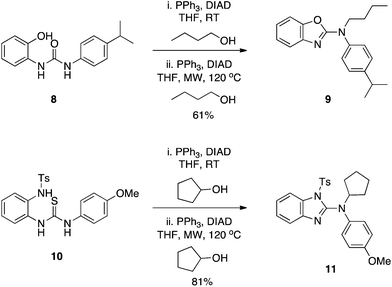 |
| | Scheme 4 2-Ureidophenols and tosyl-activated 2-thioureidoanilines undergo intramolecular Mitsunobu reactions to deliver N2-substituted benzoxazoles and benzimidazoles, respectively. | |
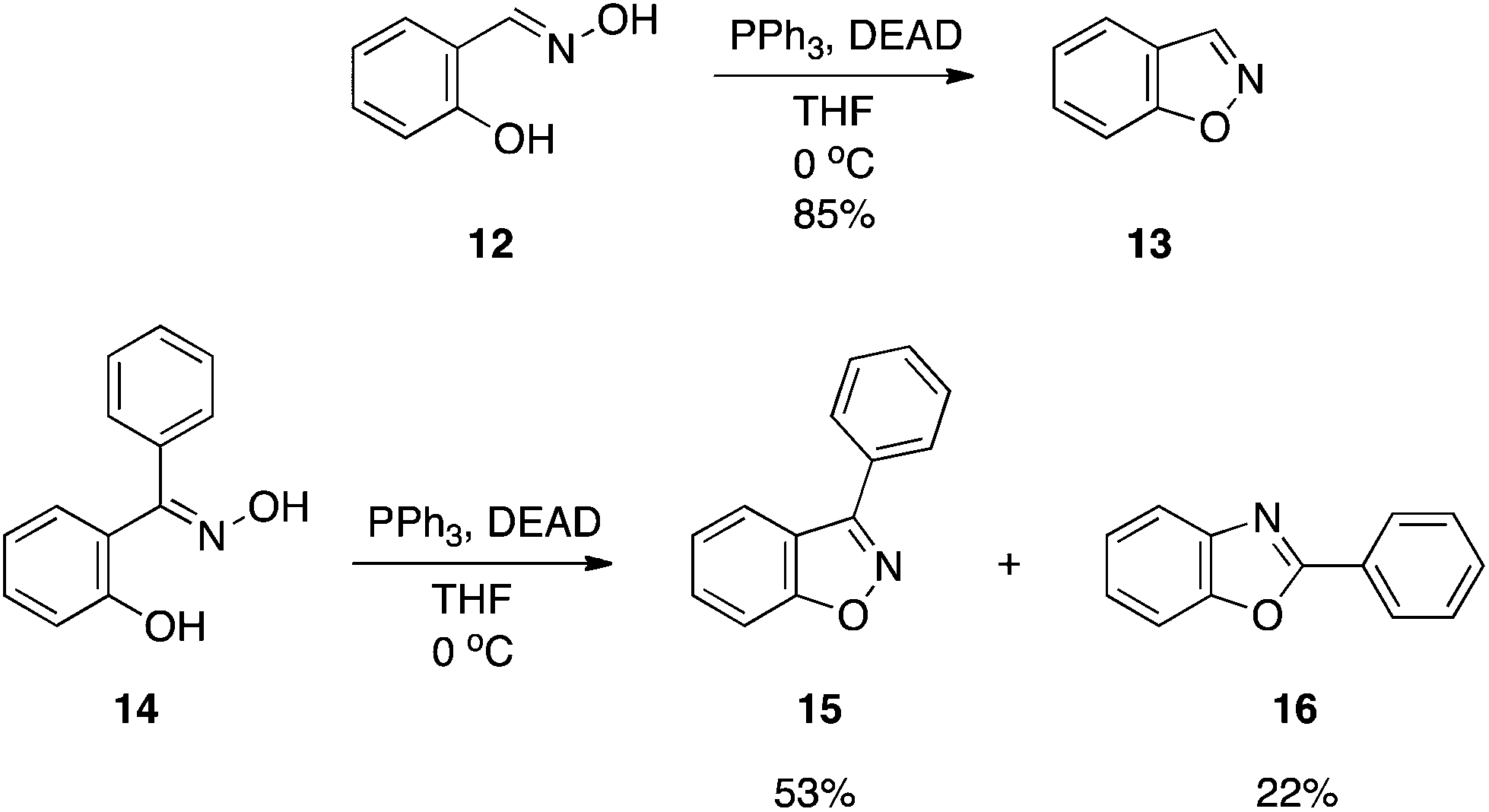
Salicylaldoximes: synthesis of 1,2-benzisoxazoles.
1,2-Benzisoxazoles are useful species for monitoring the efficiency of artificial enzyme systems since the presence of a general base and an appropriately-positioned hydrogen bond donor will catalyze a ring-opening reaction, provided there is an abstractable hydrogen at the 3-position, to afford the corresponding 2-cyanophenol (salicylonitrile).10 This β-elimination reaction is referred to as the Kemp elimination,11 and is one of the most widely studied reactions in biomimetic catalysis.10 A new synthetic route to 1,2-benzisoxazoles (e.g.13) has been accomplished by the dehydrative ring closure of salicylaldoximes (e.g.12) with DEAD–PPh3, as depicted in Scheme 5.12 It was noted that the reactions must be run dilute (1 mM) to achieve the good to excellent yields reported. Also, DEAD must be added slowly. Attempts to expand the chemistry to ortho-hydroxyarylketoximes as a means of preparing 3-substituted, 1,2-benzisoxazoles were met with mixed results. Methyl and ethyl ketoximes proved excellent substrates but the phenyl ketoxime 14 afforded the corresponding 3-phenyl-1,2-benzisoxazole 15 in only moderate yield (53%). The low yield was partially due to a Beckmann rearrangement that resulted in the isomeric 2-phenylbenzoxazole 16. Modification of the reaction conditions to reduce formation of the benzoxazole were unsuccessful.
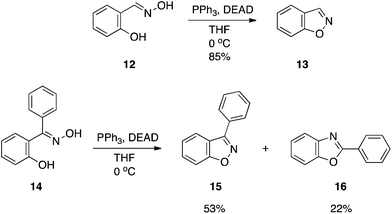 |
| | Scheme 5 The intramolecular attack of activated oximes by phenols: synthesis of 1,2-benzisoxazoles. | |
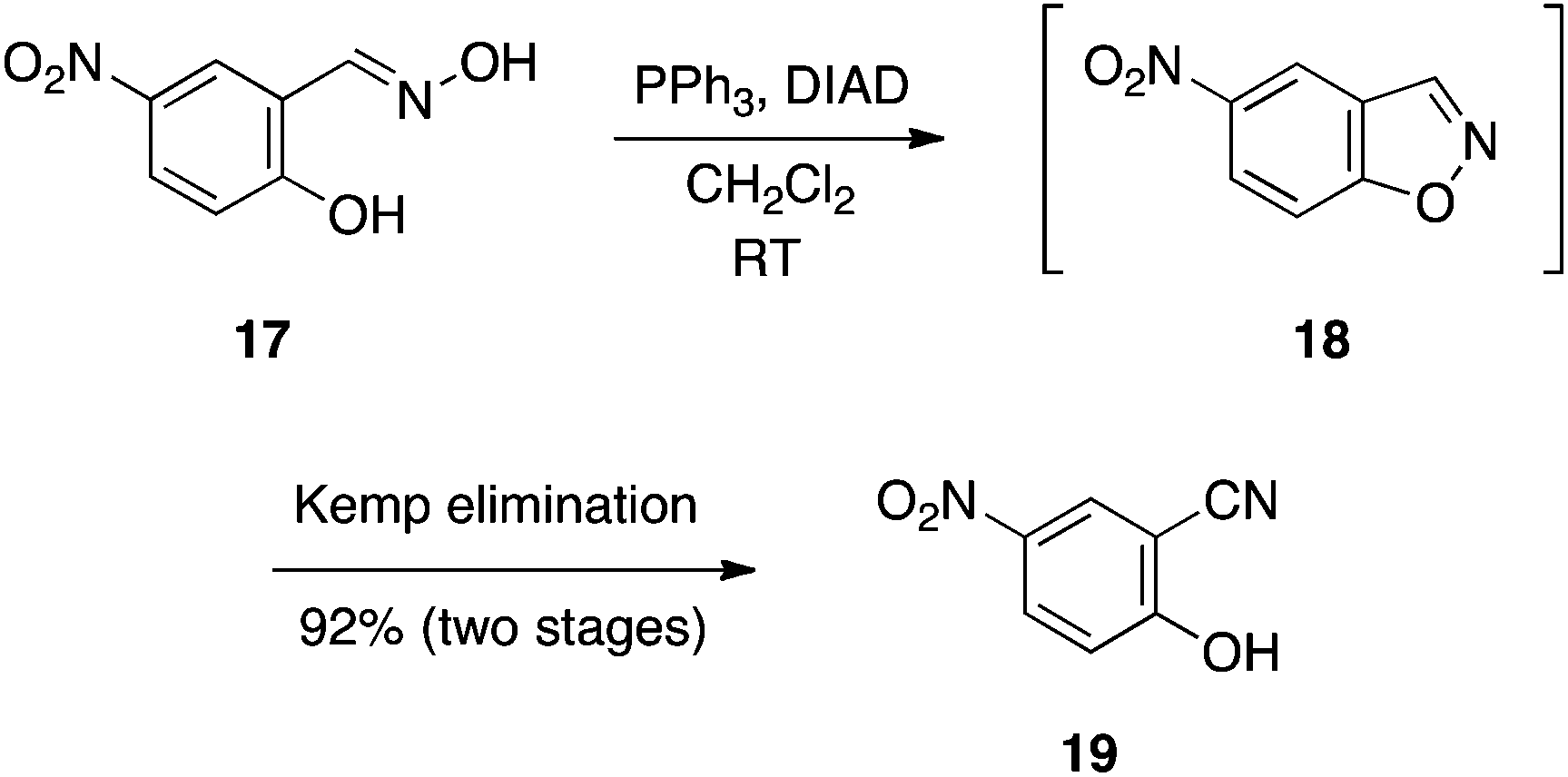
Salicylaldoximes: synthesis of salicylonitriles.
In connection with our research program to develop Mcl-1 inhibitors,13 we sought more cell penetrable bioisosteres of the carboxylic acid function in our compounds.14 Exhibiting pKa values of around 7, it was speculated that salicylonitriles might serve this role.15 We reasoned that treating salicylaldoxime with an excess of the Mitsunobu reagents would effect the expected cyclodehydration as in the previous sub-section, and then, by virtue of the basicity of the betaine, a subsequent β-elimination would ensue to afford the corresponding salicylonitrile. Rewardingly, this was found to be the case, as illustrated by the reaction of 5-nitrosalicylaldoxime (17) in Scheme 6 to generate 5-nitrosalicylonitrile (19) via the 1,2-benzisoxazole 18.16 The chemistry was compatible with a variety of functional groups, yielding the salicylonitriles in good to excellent yields. Notably, reactions were swift, typically being complete within 30 min at room temperature, and the cleanliness of the reactions coupled with the acidity of the products allowed their isolation through simple acid–base work-ups.
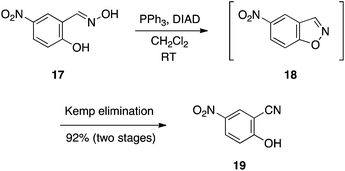 |
| | Scheme 6 One-pot synthesis of salicylonitriles: a Mitsunobu-mediated cyclodehydration–β-elimination cascade. | |
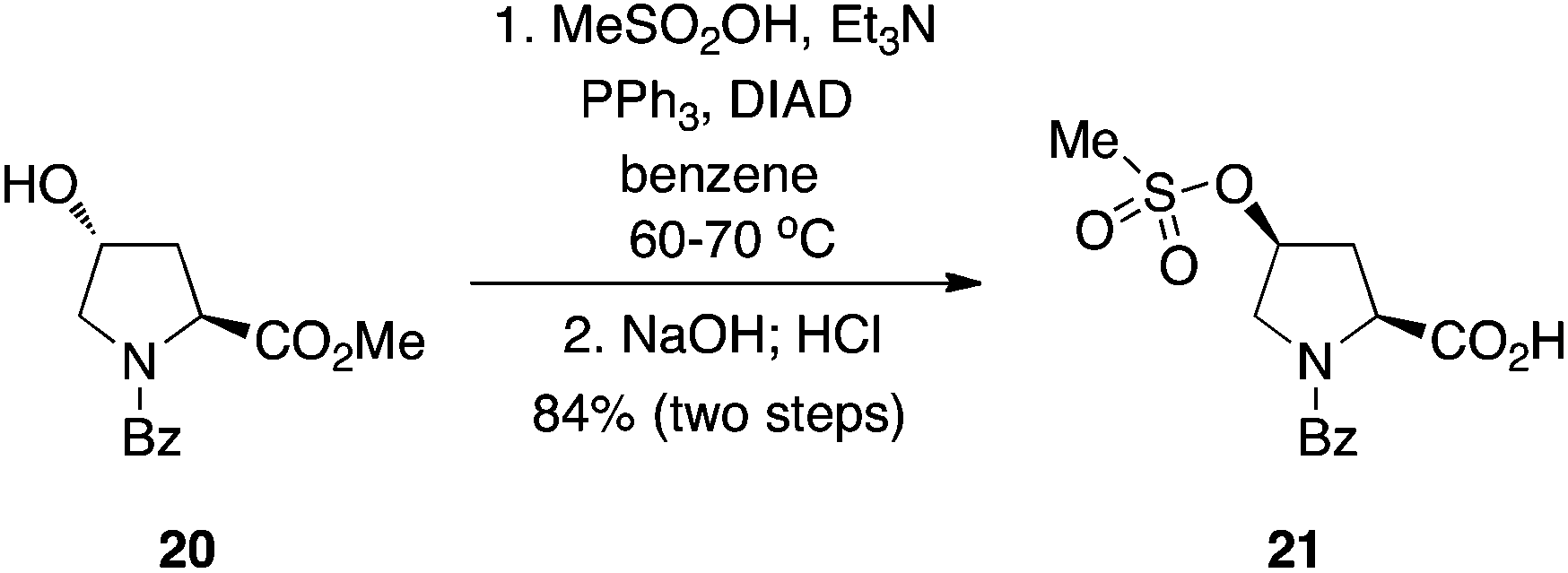
Sulfonic acids.
A group at Bristol-Myers Squibb showed that sulfonic acids are suitable pronucleophiles in the Mitsunobu reaction.17 As with carboxylic acids, the routine inversion of stereochemistry of alcoholic secondary substrates was observed (Scheme 7). The chemistry is scalable, working just as well on a multi-kilo scale.
 |
| | Scheme 7 Synthesis of a sulfonate ester employing a sulfonic acid as the pronucleophile. | |
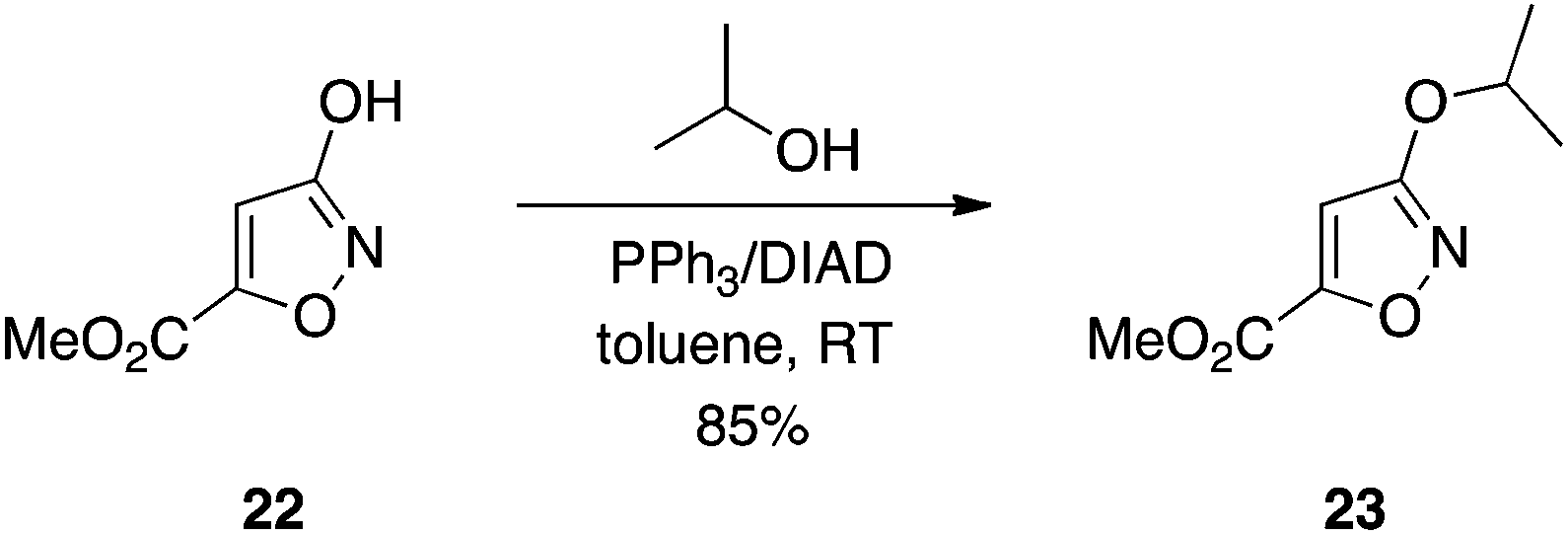
3-Hydroxyisoxazoles.
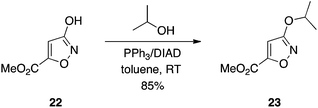
As already alluded to, carboxylic acids, as their charged carboxylates, often limit the cellular activity of drug molecules through inhibiting cell penetration. More lipophilic bioisosteres, such as tetrazoles, have served as effective surrogates of carboxylic acids that aid in cell entry.14 The 3-hydroxyisoxazole motif is also bioisosteric with the carboxylic acid function.18 What is especially noteworthy about 3-hydroxyisoxazoles is, like the acyl sulfonamides, the acidic group can be functionalized bilaterally.19 This is in contrast to carboxylic acids and tetrazoles where the acidic group occurs at the terminus of a drug molecule. Chemical transformation of the 3-hydroxyisoxazole scaffold, for example in protecting group strategies, is hampered by its ambidentate nature, and classical alkylation leads to a mixture of N and O functionalizations.20 We have demonstrated that Mitsunobu alkylation of 3-hydroxyisoxazoles predominantly yields the O-alkylated products (≥95% O-selectivity in many cases); an example is given in Scheme 8.21 Moreover, the Mitsunobu reaction is a more convenient means to functionalize the motif than the classical alkylation procedure: reactions proceeded quickly at room temperature, rather than requiring heat for several days.19 Also, in the context of methylation, this chemistry avoids the use of explosive diazomethane.
 |
| | Scheme 8 Regioselective O-alkylation of 3-hydroxyisoxazoles under Mitsunobu control. | |
Nitrogen pronucleophiles
Sulfonamides, exhibiting pKa values of about 10, are excellent nucleophiles in the Mitsunobu reaction.2–5 Azide is also a superb nucleophile and may be administered in a variety of forms: hydrazoic acid, metal azide or diphenylphosphoryl azide (DPPA).2 Care must be taken, however, as an excess of PPh3 might result in transformation of the organic azide into the corresponding iminophosphorane, which constitutes the first step of the Staudinger reaction to generate primary amines.
Phthalimide: synthesis of primary amines.
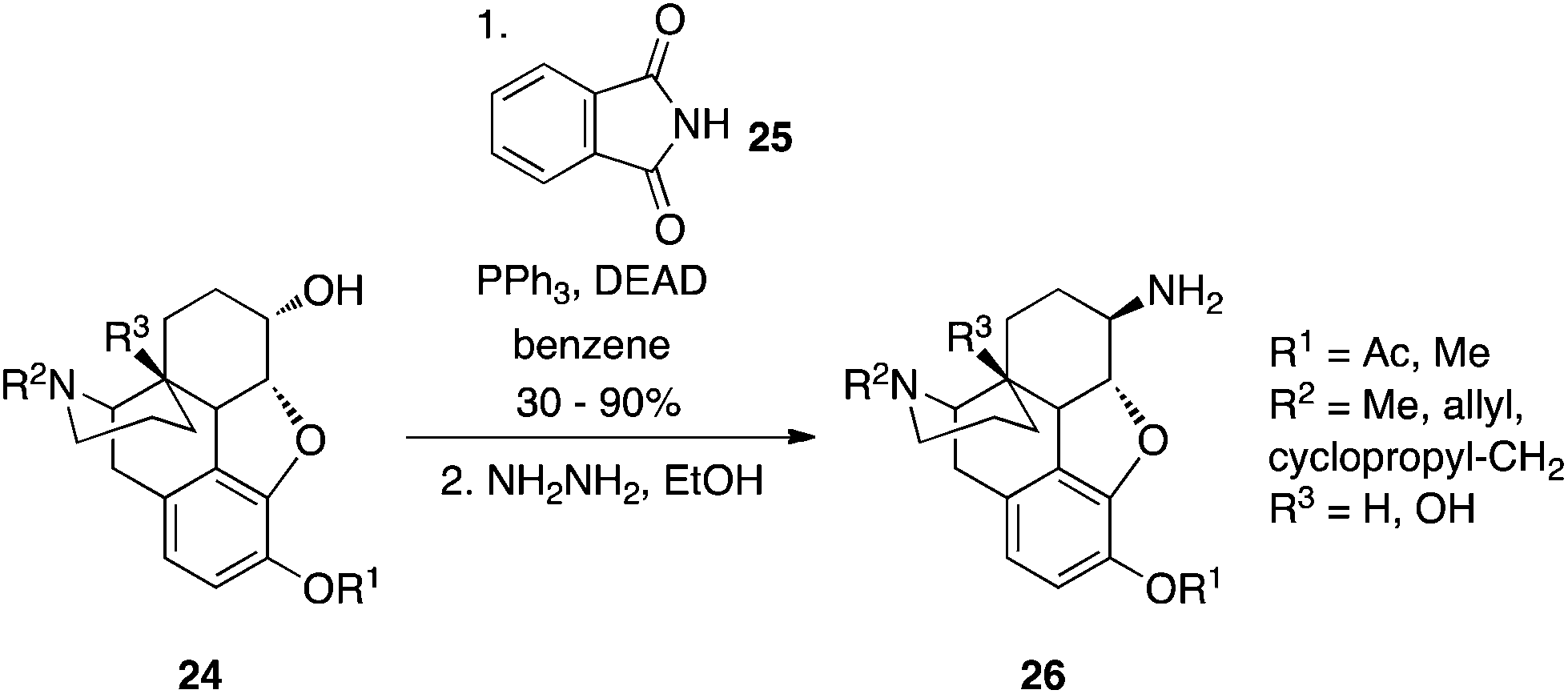
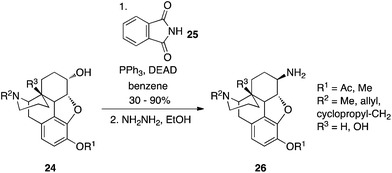
Protected primary amines can be synthesized from alcohols using phthalimide (25) as the pronucleophile.22 Reminiscent of the Gabriel synthesis, the resulting N-alkylated phthalimide is then subjected to hydrazinolysis to reveal the free primary amine. An example of this chemistry in action is shown in Scheme 9 in which β-naltrexamine derivatives were prepared from dihydrocodeine.
 |
| | Scheme 9 Phthalimide in the Mitsunobu reaction permits a two-step conversion of alcohols to primary amines. | |
Nitrobenzenesulfonamides: synthesis of secondary amines.
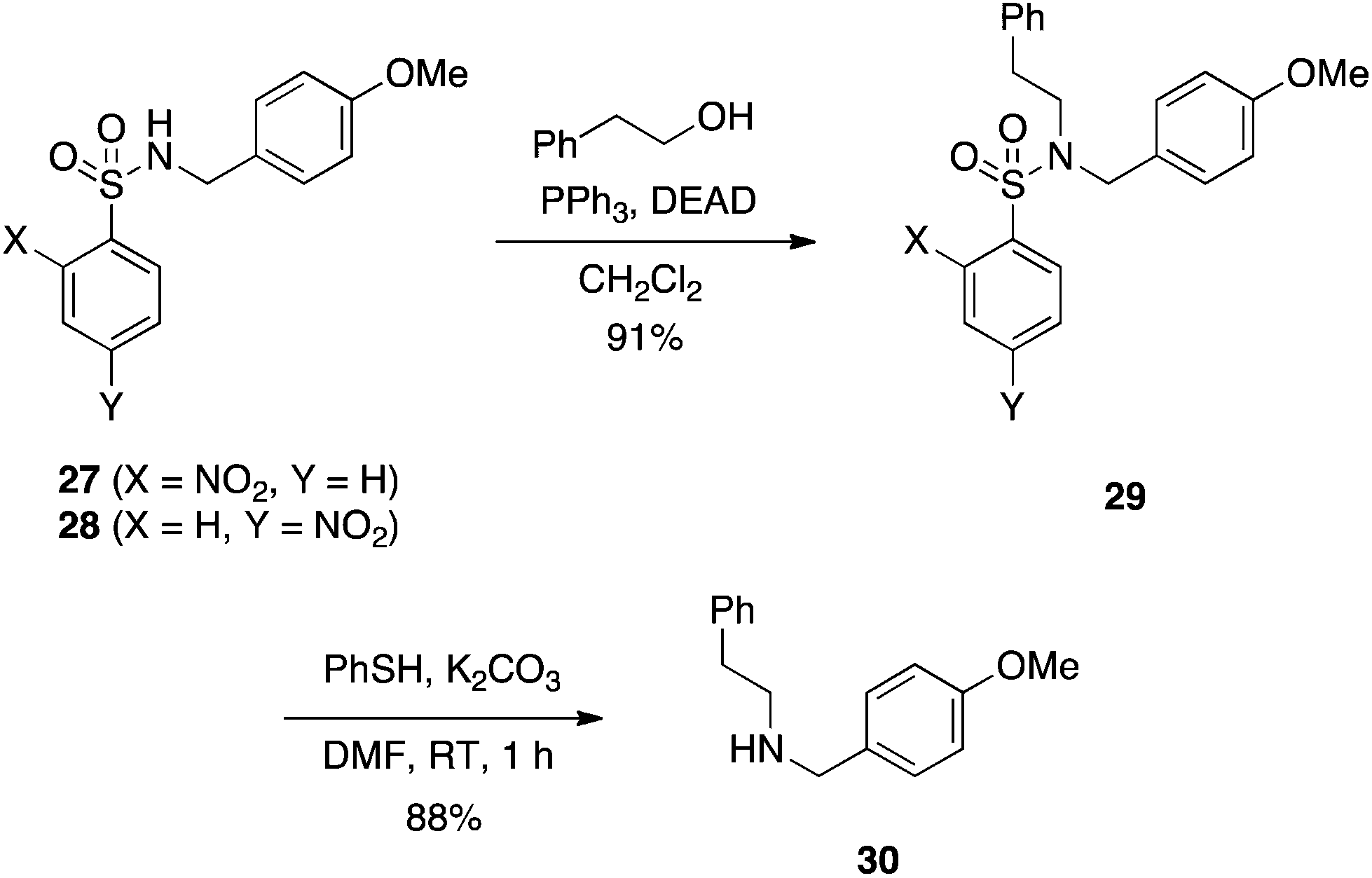
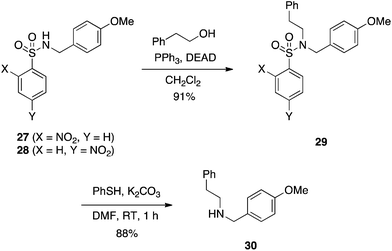
In 1989, Weinreb reported that protected sulfonamides such as BocNHTs are proficient pronucleophiles in the Mitsunobu reaction, in concordance with their acidic pKas of around 5.23 Sequential removal of the Boc protecting group followed by another Mitsunobu reaction permits the synthesis of tertiary sulfonamides, while concomitant removal of both protecting and sulfonyl groups reveals the primary amine.23 In a similar vein to Weinreb's work, Fukuyama developed a strategy to synthesize secondary amines through the use of nitrobenzenesulfonamides (e.g.27 and 28) as both the activating group and protecting group. The first step is a Mitsunobu reaction on the sulfonamide, and the second step is a deprotection of the sulfonyl moiety. Colloquially, this reaction is referred to as the Fukuyama–Mitsunobu reaction, and an example is given in Scheme 10.24 Yields are typically very good to excellent for the Mitsunobu step. When ortho- or para-nitrobenzenesulfonamides (Ns) are employed, the sulfonyl group can be removed by treatment with thiophenol and K2CO3 for 1 h to deliver the secondary amines in very good yields. Use of ortho,para-dinitrobenzenesulfonamide (DNs) in place of Ns as the activating/protecting group is also effective and, due to its greater lability, the DNs group may be deprotected selectively over the Ns group by stirring with thioglycolic acid in a mildly basic medium.25 Notably, the resulting by-product carrying a carboxylic acid can be removed by a basic wash of the ethereal layer during reaction work-up. Fukuyama later applied his Ns protection/activation strategy to the synthesis of some of the iboga alkaloids.26
 |
| | Scheme 10 Fukuyama-Mitsunobu synthesis of secondary amines employing thiolate-labile Ns protecting/activating groups. | |
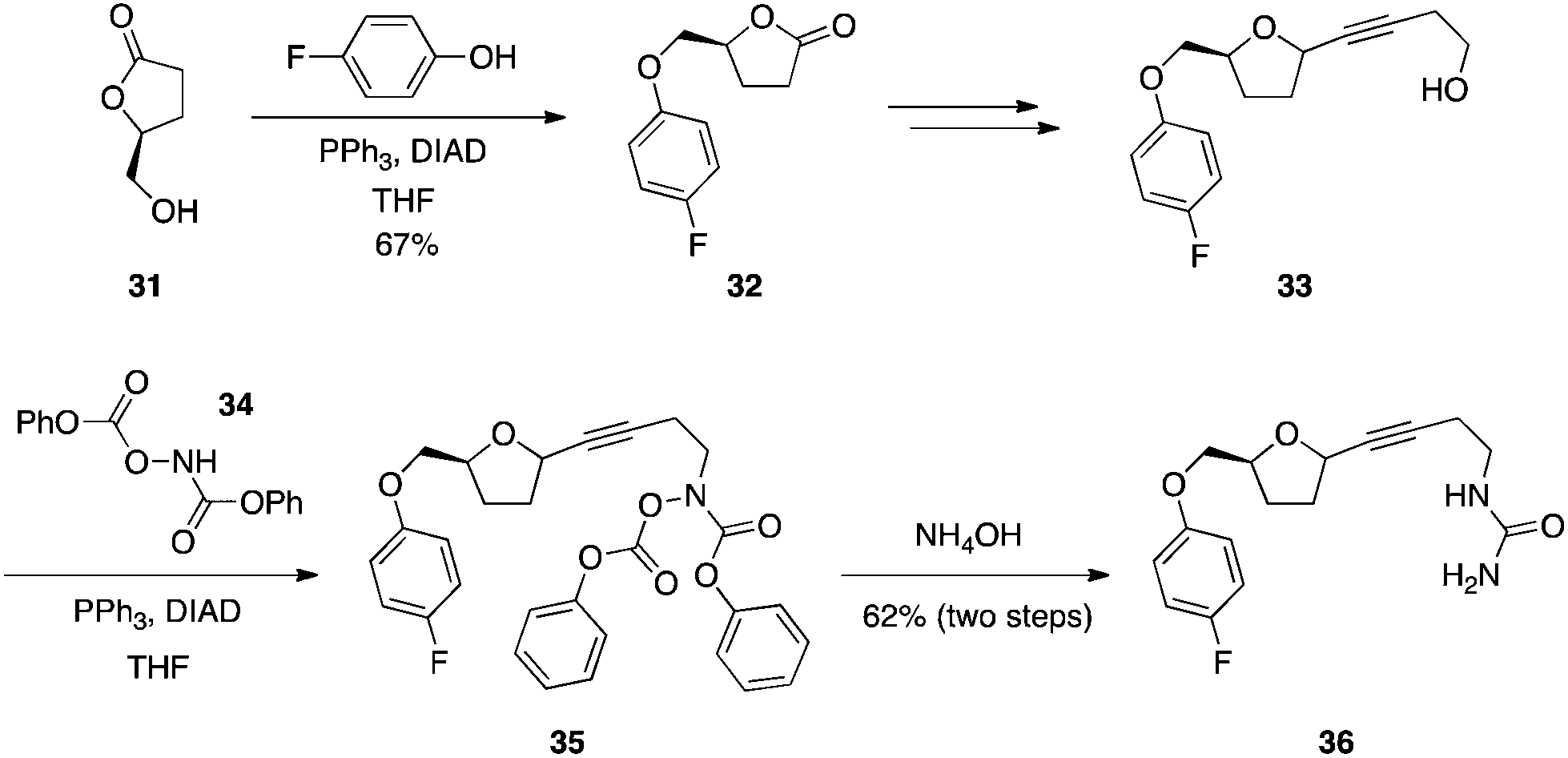
Activated hydroxylamines: synthesis of ureas.
Acylated hydroxylamines are sufficiently acidic to couple to alcohols.28 In the total synthesis of the 5-lipoxygenase inhibitor CMI-977 (36) (Scheme 11), the alcohol of lactone 31 underwent a Mitsunobu reaction with 4-fluorophenol to deliver ether 32. After further synthetic transformations, resulting molecule 33 was subjected to a second Mitsunobu reaction with N,O-bis(phenoxycarbonyl)hydroxylamine (34), which, upon aminolysis, furnished CMI-977.27
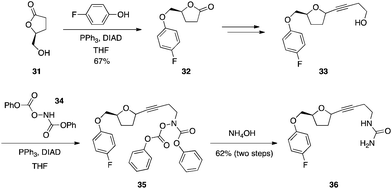 |
| | Scheme 11 Synthesis of ureas with activated hydroxylamines. | |
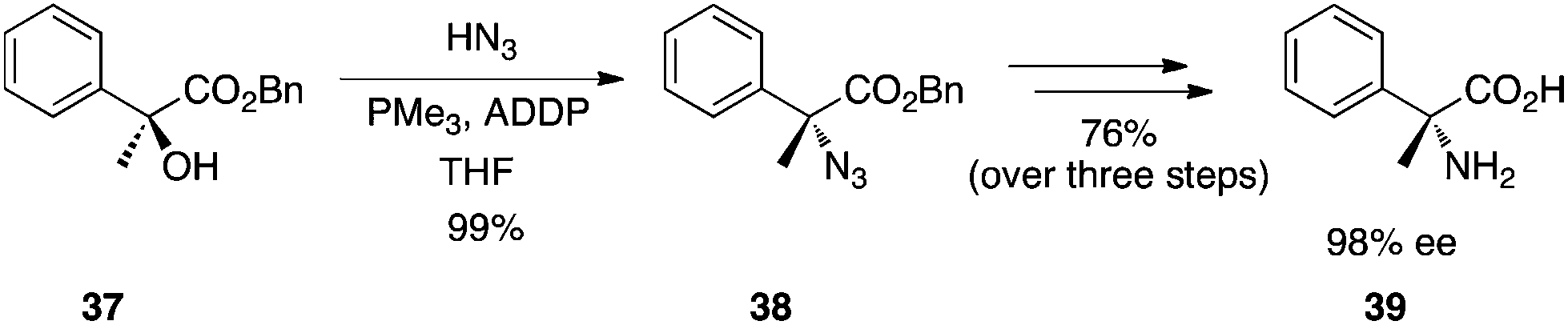
Synthesis of optically active α,α-disubstituted amino acids.
Chiral tertiary α-hydroxy esters can be converted to α-azido esters with hydrazoic acid as the pronucleophile.28 As complete inversion of the stereochemical configuration is observed, this chemistry provides, after reduction and ester hydrolysis, an efficient and reliable strategy to synthesize α,α-disubstituted amino acids. An example of this chemistry is depicted in Scheme 12. However, it is unclear how scalable is the chemistry for several reasons. First, hydrazoic acid is toxic and potentially explosive. Second, reaction optimization revealed 1,1′-(azodicarbonyl)dipiperidine (ADDP) as the preferred azodicarbonyl species but ADDP is relatively costly and is required in excess. Finally, PMe3 is necessary as the phosphine partner of the redox system. Notwithstanding its stench, PMe3 is pyrophoric and must be handled with extreme care. Nevertheless, the total inversion of the tertiary alcohol's chiral centre renders this chemistry a powerful tool in the synthesis of hindered amino acids, at least on a laboratory scale.
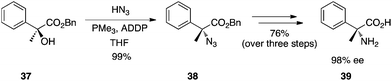 |
| | Scheme 12 Synthesis of an α,α-disubstituted amino acid from tertiary α-hydroxy esters. | |
Activated aminoazoles.
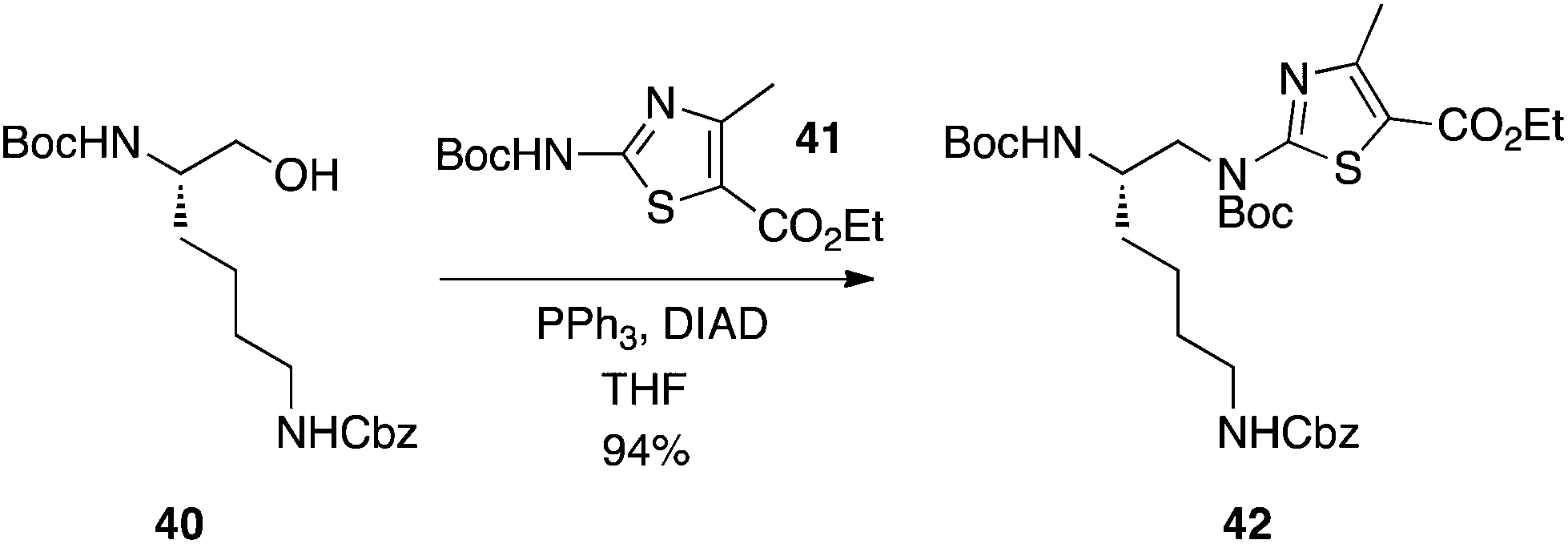
Kim and Kahn from Molecumetics Ltd have demonstrated that Boc-protected 2-aminoazoles, such as 41, are sufficiently activated to smoothly undergo the Mitsunobu reaction to furnish some lysine and arginine analogues (Scheme 13).29 Given their high yields, it appears that there was negligible interference, if any, by the aliphatic Boc and Cbz carbamates, which is consistent with their higher pKa values of around 16–18.
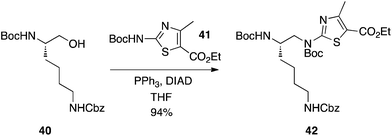 |
| | Scheme 13 Alkylation of a Boc-protected/activated 2-aminoazole. | |
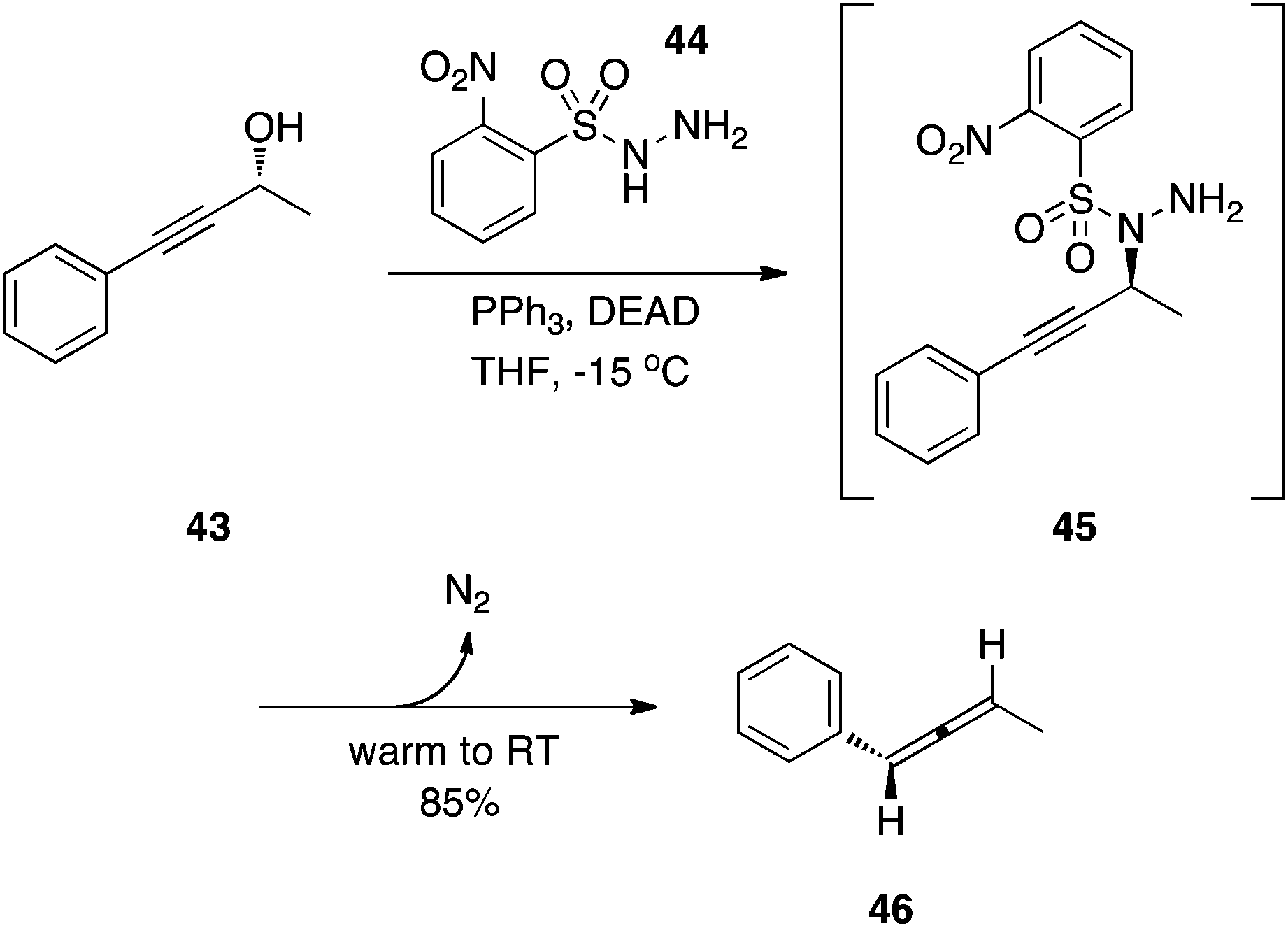
2-Nitrobenzenesulfonylhydrazine: synthesis of allenes.
The stereospecific synthesis of allenes has been accomplished by Myers and colleagues.30 As shown in Scheme 14, the Mitsunobu amination of a propargyl alcohol with the sulfonyl hydrazide NsNHNH2 (44) afforded the expected product 45, which was not isolated. Upon allowing the reaction mixture to warm to RT, N2 was lost as was the arylsulfinic acid to generate allene 46.
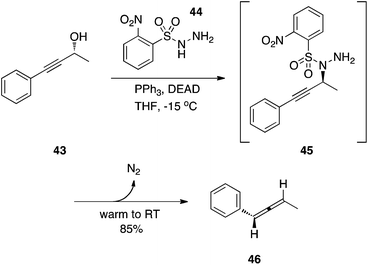 |
| | Scheme 14 Stereospecific synthesis of an allene. | |
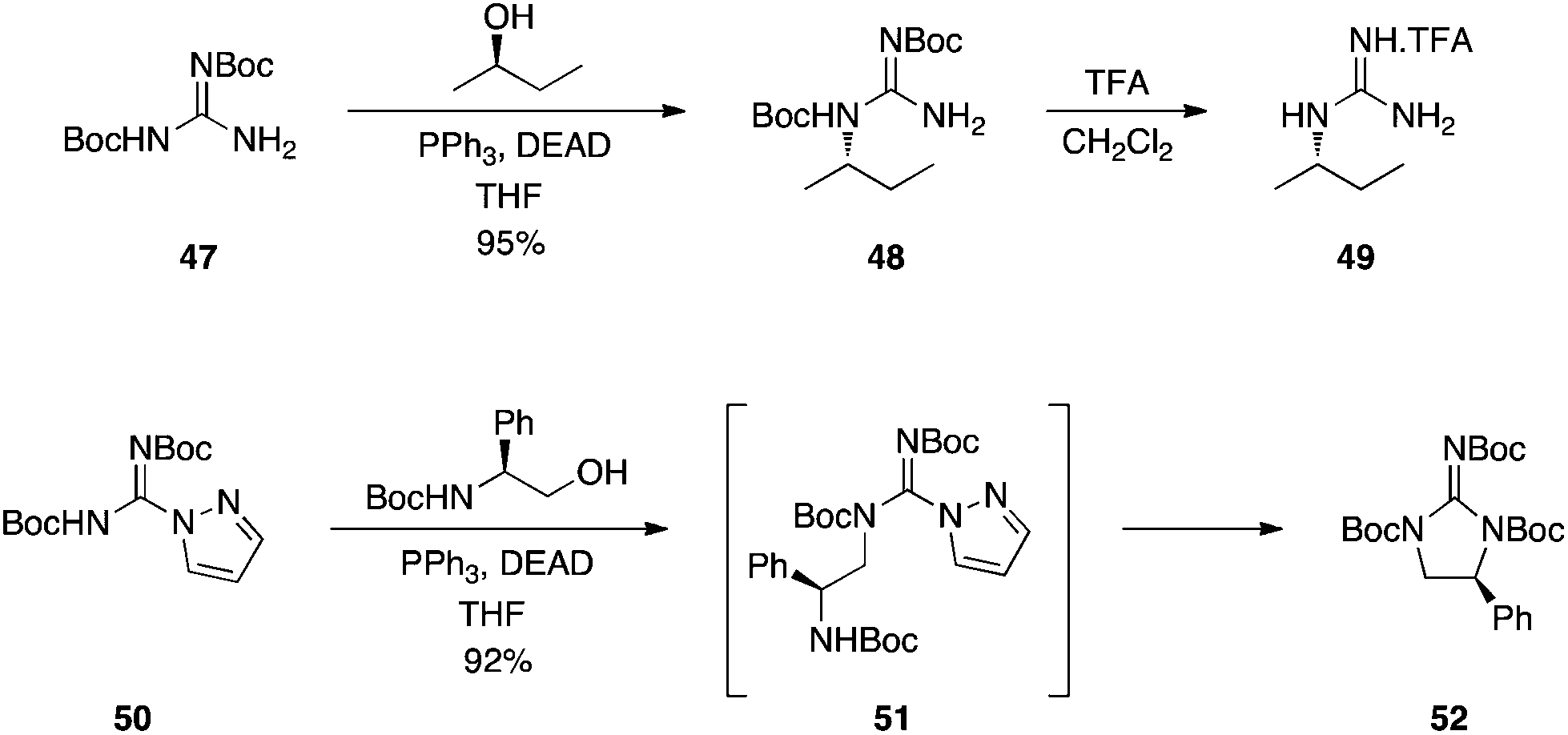
Activated guanidines.
Kozikowski and Dodd showed that bis-Boc-protected guanidine 47, as well as bis-Cbz-protected guanidine, coupled in excellent yields to a variety of alcohols, delivering, after deprotection, mono-substituted guanidines, such as 49 (Scheme 15, upper panel).31 As expected, the reactions proceeded with inversion of configuration. Later, Kim and colleagues at Molecumetics, Ltd. demonstrated that similarly activated guanylating agents, such as 50, are also capable of coupling to primary and secondary alcohols under Mitsunobu conditions.32 Subsequent reaction with an amine then affords disubstituted guanidines. When suitable alcohols are used, spontaneous cyclization occurs, as shown in Scheme 15 (lower panel).
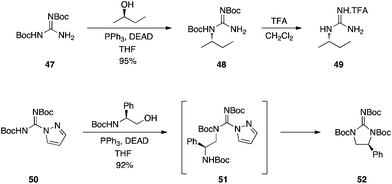 |
| | Scheme 15 Synthesis of substituted guanidines exploiting the Boc protecting group as an activating group. | |
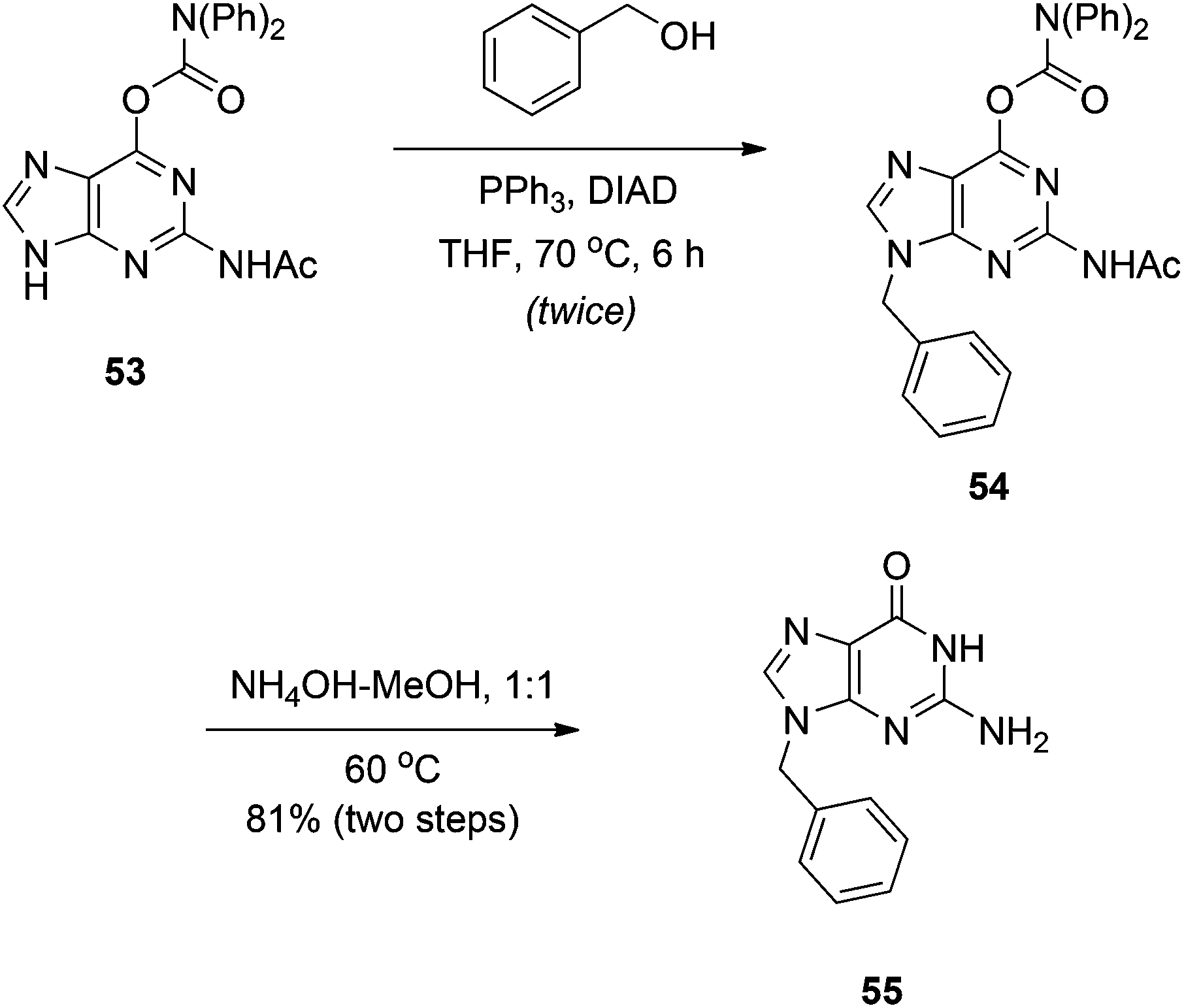
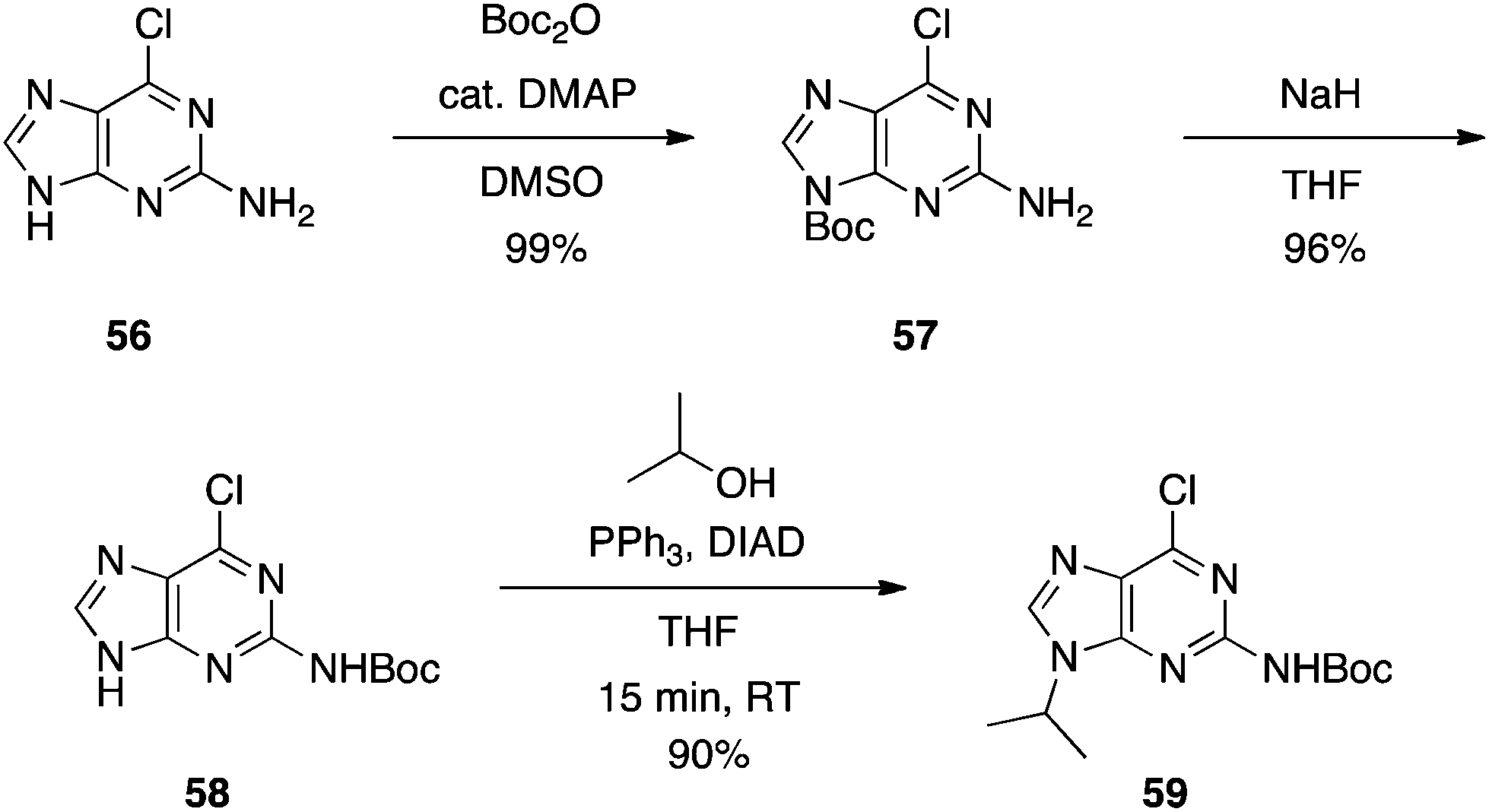
Purines.
N
9-Regioselective alkylation of 2-amino-6-chloropurine using Mitsunobu chemistry was first reported in 2005, although the substrate scope was limited and yields were poor to modest.33 Shi and colleagues were keen to expand on this work to aid in their discovery of self-assembled nucleoside molecular architectures.34 In particular, the authors focused on the guanine precursor 53. Like guanine, O-carbamate-N-acetate protected guanine 53 proved only partially soluble in THF and so, to promote solubility, the Mitsunobu reaction was run at 70 °C. Good to excellent yields of the N9 product were reported, which was confirmed by X-ray crystallography, but the high temperature necessitated two rounds of addition of the Mitsunobu reagents due to their decomposition in the reaction.34 Deprotection of the products with NH3 in MeOH delivered the desired N9-substituted guanines (Scheme 16). It is interesting to note that no alkylation at the N2 position of the NHAc group was reported, even at 70 °C, because chemists at Utah showed that this is achievable using the DIAD-triisopropyl phosphite redox system at 50 °C.35
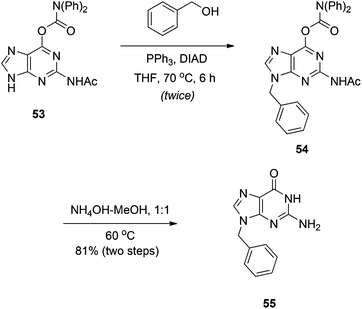 |
| | Scheme 16
N
9-Regioselective alkylation of the guanine precursor 53. | |
This chemistry is also compatible with a variety of other purine bases, primary and secondary alcohols, and X-ray crystal structures confirmed complete inversion of stereochemistry. Overall, Shi and colleagues introduced a reproducible and high-yielding approach to effect the N9-regioselective alkylation of purines,34 in contrast to the classical alkylation strategy of R-Hal/K2CO3 in DMF that often delivers an approximate 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of N9:N7 products.36
:1 mixture of N9:N7 products.36
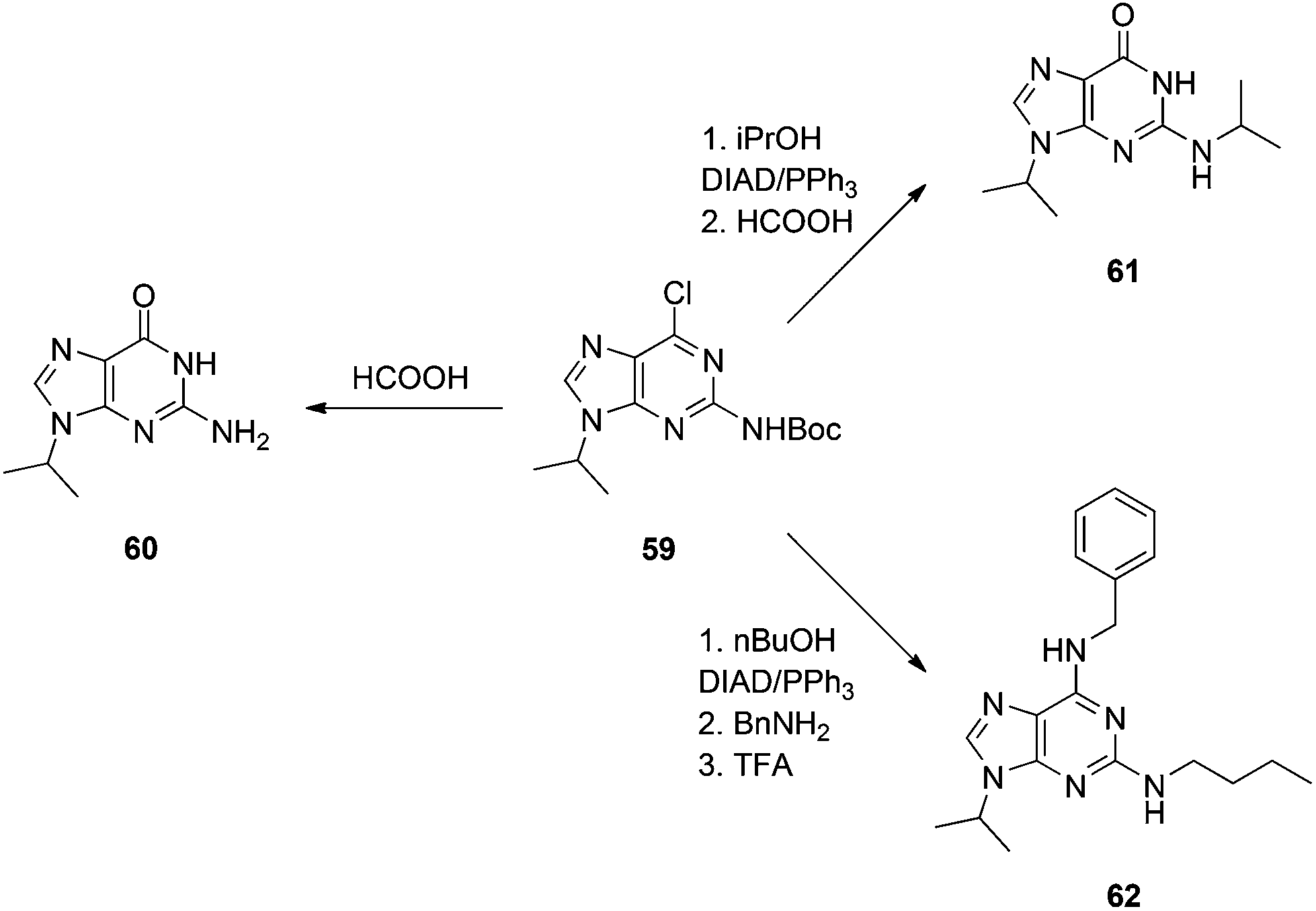
We also required an efficient means of synthesizing N9-alkylated guanines towards some novel PNA molecules, and considered that Shi's work might be improved upon by Boc-protecting the N2 position of 2-amino-6-chloropurine. It was predicted this would greatly enhance the solubility of the purine in THF, allowing the Mitsunobu reaction to proceed at room temperature. Moreover, the greater steric bulk of the Boc group versus Ac was postulated to offset any undesired N2-alkylation that might have been experienced in Shi's chemistry. Accordingly, N2-Boc-2-amino-6-chloropurine (58) was prepared and then exposed to Mitsunobu conditions: 58 coupled very efficiently to various primary and secondary alcohols to deliver the N9-alkylated products in good to excellent yields at room temperature within 15 min (Scheme 17), in stark contrast to Shi's conditions (Scheme 16), and we observed no N2-alkylation.37 The products were then subjected to heat in formic acid to effect Boc group deprotection and hydrolysis of the 6-chloro group in one pot, furnishing the N9-alkylated guanines (e.g.60, Scheme 18). Note: since our original report of this chemistry, we have found the nature of the NaH to be critical in the Boc transfer reaction from 57 to 58 – NaH in mineral oil provides capricious results, occasionally with little to no Boc transfer, whilst dry NaH consistently drives complete transfer. We extended this chemistry further and showed that the N2 position could be alkylated under modified Mitsunobu conditions, which could then undergo treatment with formic acid to afford N2,N9-disubstituted guanines (e.g.61), or nucleophilic aromatic substitution with various amines followed by treatment with TFA to yield N2,N6,N9-tri-substituted purines, including the CDK inhibitor bohemine (62) (Scheme 18).37,38
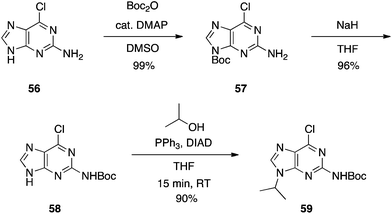 |
| | Scheme 17
N
9-Regioselective alkylation of an alternative and more soluble guanine precursor than 53. | |
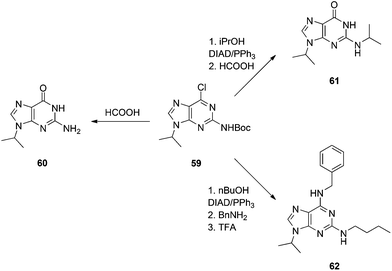 |
| | Scheme 18 The versatility of N9-substituted-N2-Boc-2-amino-6-chloropurine. | |
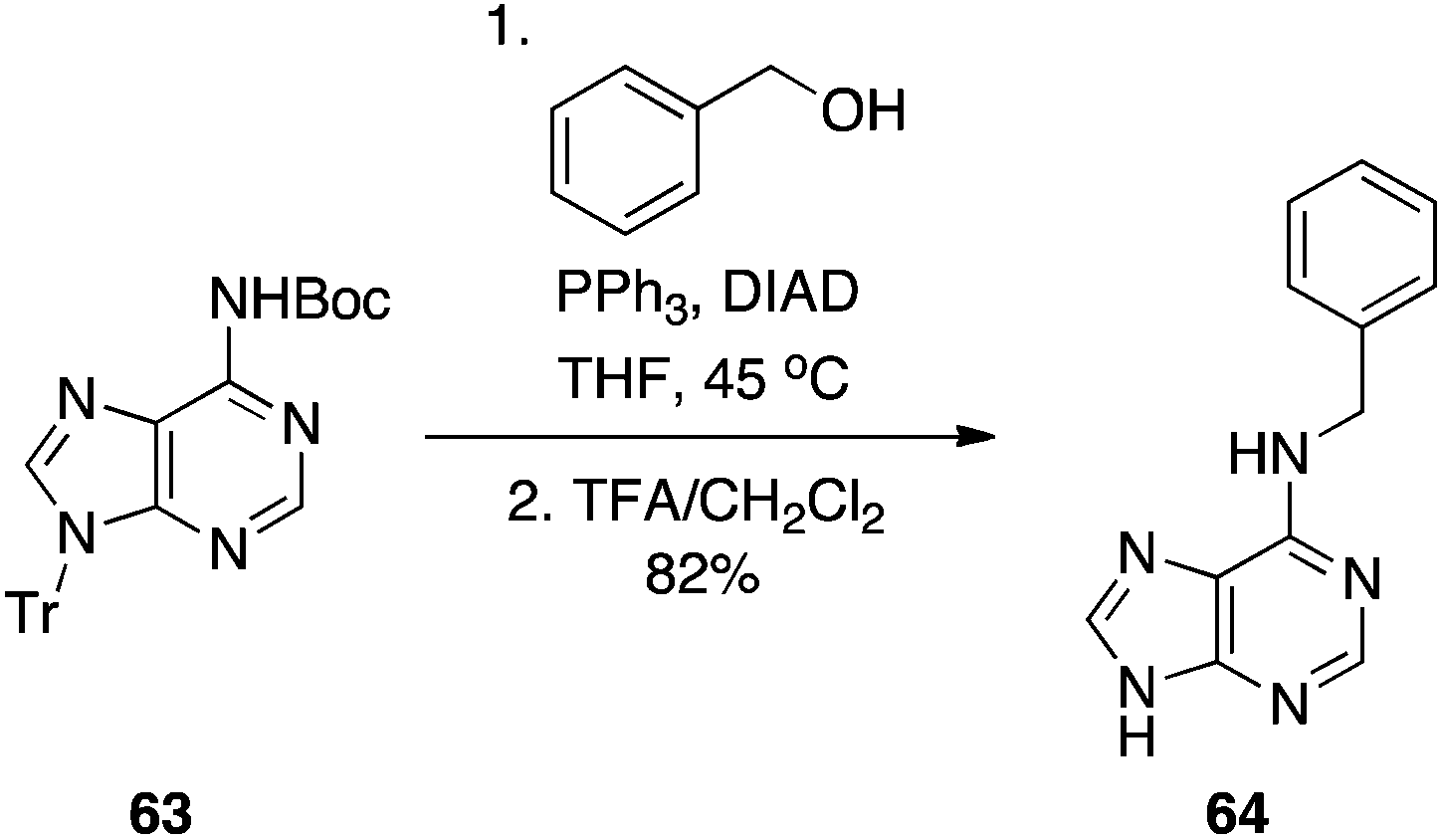
Later, we similarly activated the N6 position of adenine and adenosines with a Boc group (63), and demonstrated it, too, is an effective pronucleophile in the Mitsunobu reaction, providing a milder alternative to the traditional nucleophilic aromatic substitution that is employed to functionalize the 6-position of the purine nucleus (Scheme 19).39 In summary, the practicality, efficiency and adaptability (guanines as well as other purines may be synthesized) suggest this chemistry will be of great use in the regioselective functionalization of nucleobases.
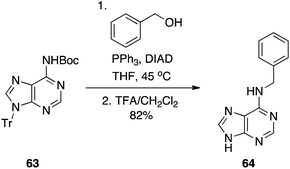 |
| | Scheme 19 Alkylation of the exocyclic N6-amino group of adenine. | |
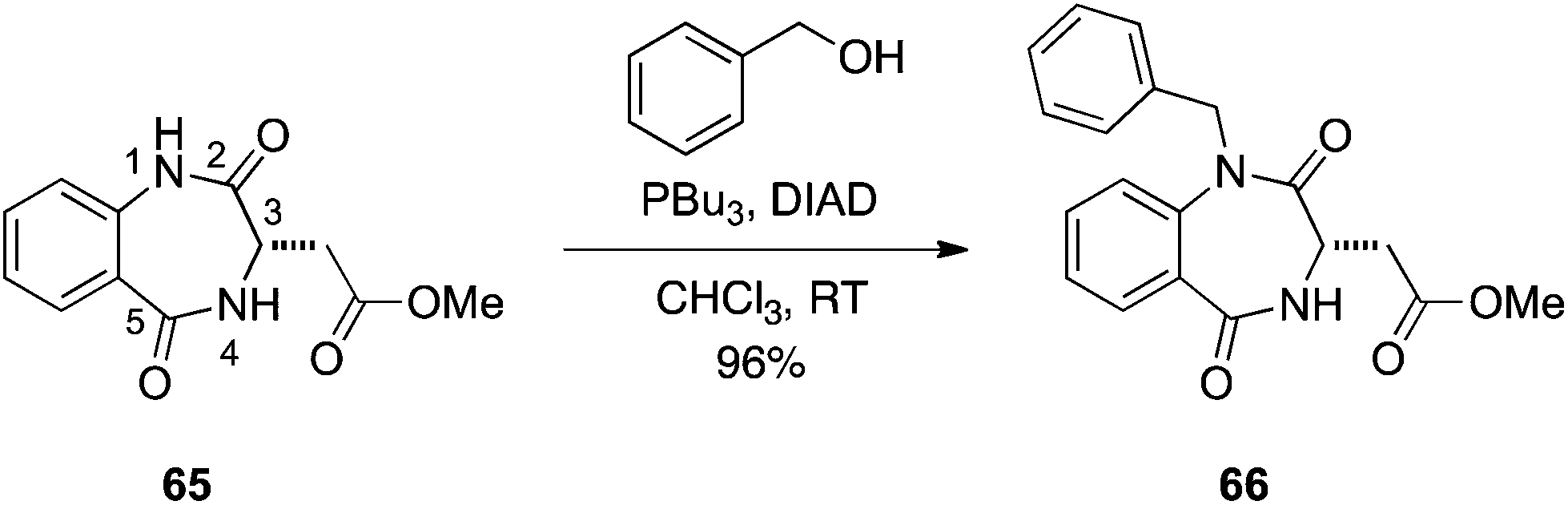
Benzodiazepine-2,5-diones.
Given their roles in proteomimicry,40,41 coupled with our interest in this area of research to develop new inhibitors of protein–protein interactions, we asked if Mitsunobu chemistry could be used to regio- and chemoselectively alkylate benzodiazepine-2,5-diones (and benzodiazepine-2-ones). Classical alkylation of benzodiazepine-2,5-diones can lead to a mixture of N- and O-alkylations of the anilide function, and an excess of reagents can also result in alkylation of the benzamide group.42 Given that K2CO3 is sufficiently basic for such alkylations, we rationalized that the anilide NH group likely exhibits a pKa of around 10 and should, therefore, participate in the Mitsunobu reaction. Accordingly, we subjected benzodiazepine-2,5-dione 65 to an excess of DIAD, PPh3 and BnOH. Gratifyingly, this resulted in alkylation of the anilide functional group with no detectable alkylation of the benzamide moiety (Scheme 20).43 We conducted a solvent, phosphine and temperature study to identify the optimum conditions for N-alkylation of the anilide functional group. It was discovered that DIAD and PBu3 in CHCl3 at room temperature afforded the highest ratios of N-alkylation with excellent yields (≥95%) observed with primary alcohols. Isopropanol required heating to 40 °C to consume all of the benzodiazepine-2,5-dione, delivering the N-isopropyl product in a yield of 85% and a selectivity of 6.1:1, N:O. This chemistry was also successfully extended to the selective N-alkylation of a 1,4-benzodiazepin-2-one.43
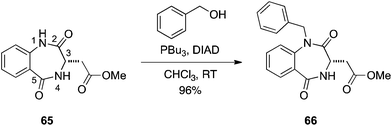 |
| | Scheme 20 Regio- and chemoselective N1-alkylation of benzodiazepine-2,5-diones. | |
Activated hydrazones.
Chemists at Johnson and Johnson investigated the reactivity of tosyl- and Boc-hydrazones in the Mitsunobu reaction.44 Exhibiting a sulfonamide-type acidic NH group, tosyl hydrazones proved excellent substrates, coupling to primary, secondary, aliphatic and benzylic alcohols in mostly very good to excellent yields (Scheme 21). On the other hand, Boc hydrazones were unreactive in the absence of electron-withdrawing groups to lower the pKa of the BocNH group.
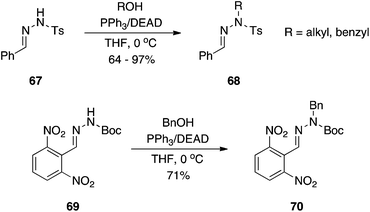 |
| | Scheme 21 Alkylation of Ts and Boc hydrazides. | |
Carbon pronucleophiles
Carbon–carbon bond forming reactions are highly sought after in the synthesis of natural products and pharmaceuticals, and new strategies to fashion such connections could present a paradigm shift in traditional drug retrosynthesis.45 There are limited reports of utilizing Mitsunobu chemistry to effect such transformations; the relatively high pKa of carbon acids is largely responsible for this.46 Falck and Manna were the first to show that this chemistry is possible by using lithium cyanide to displace an activated alcohol, which, thereby, delivered the corresponding organic nitrile.47 In more recent years, the Mitsunobu reaction has played a bigger role in fashioning carbon–carbon bonds.
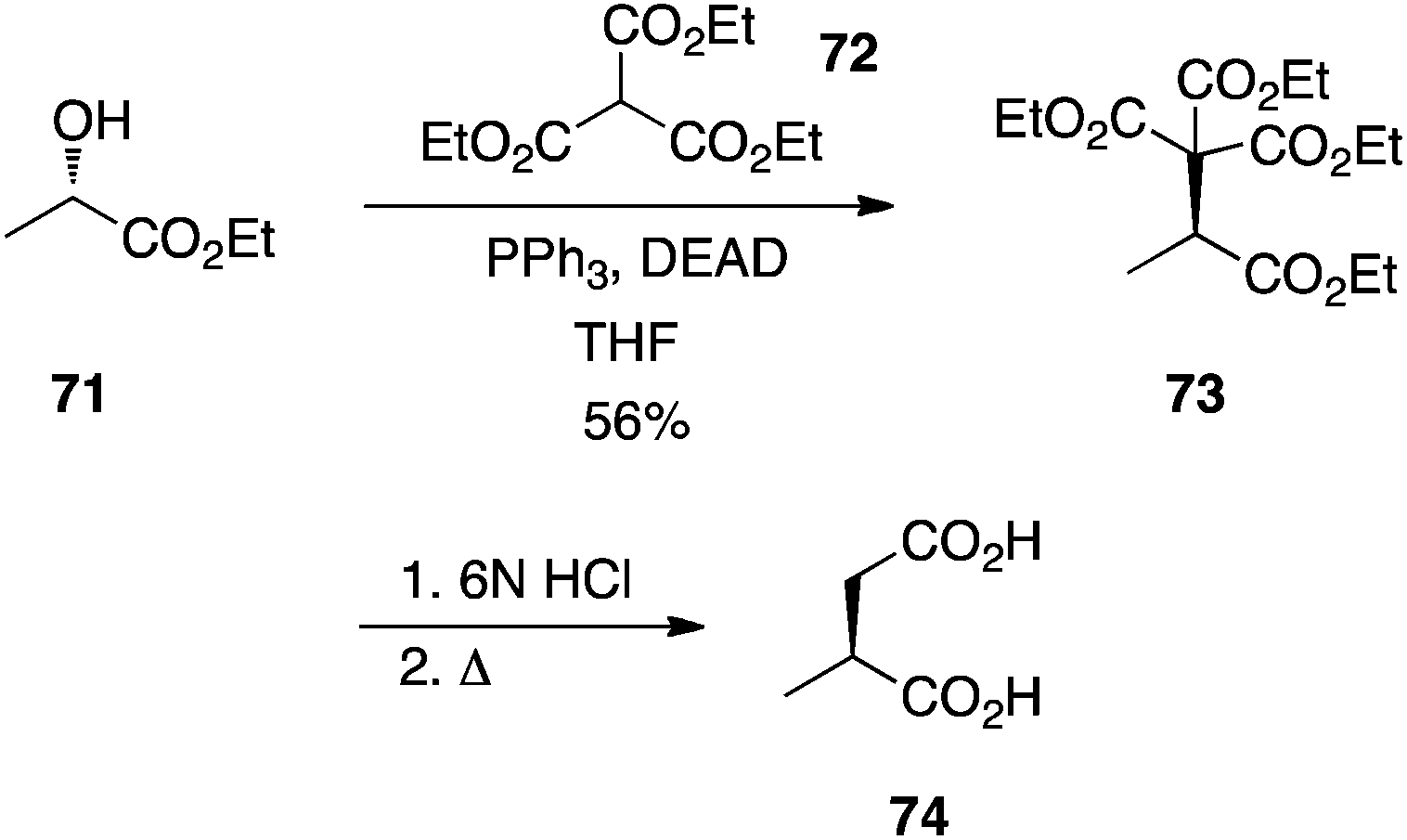
Triethyl methanetricarboxylate.
Palmisano's laboratory reported that triethyl methanetricarboxylate (72) is adequately acidic to couple to alcohols under Mitsunobu conditions with no erosion of optical purity.48 For example, (S)-2-methylsuccinic acid (74) was prepared by alkylation of (S)-ethyl lactate (71) followed by acidic hydrolysis and decarboxylation (Scheme 22).
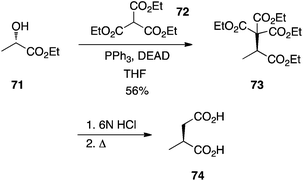 |
| | Scheme 22 Carbon–carbon bond formation in the Mitsunobu reaction with the triply-activated methine of triethyl methanetricarboxylate (72). | |
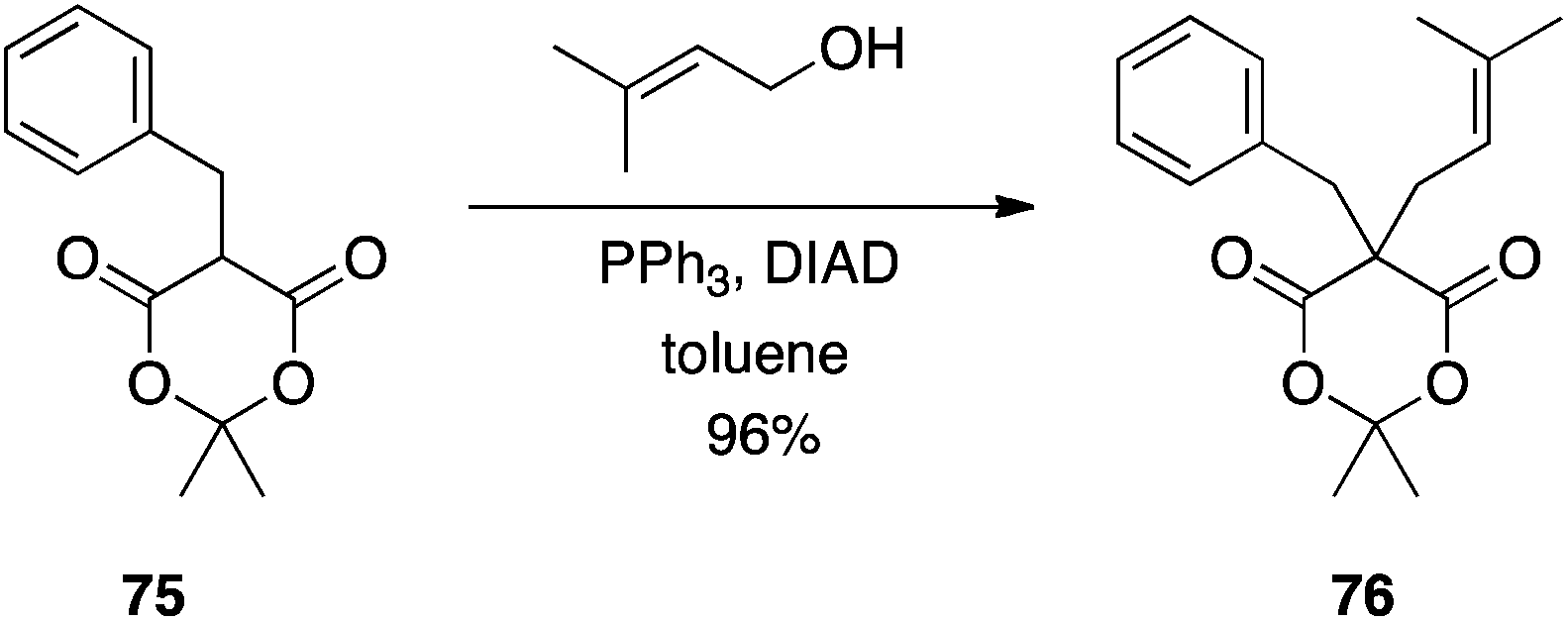
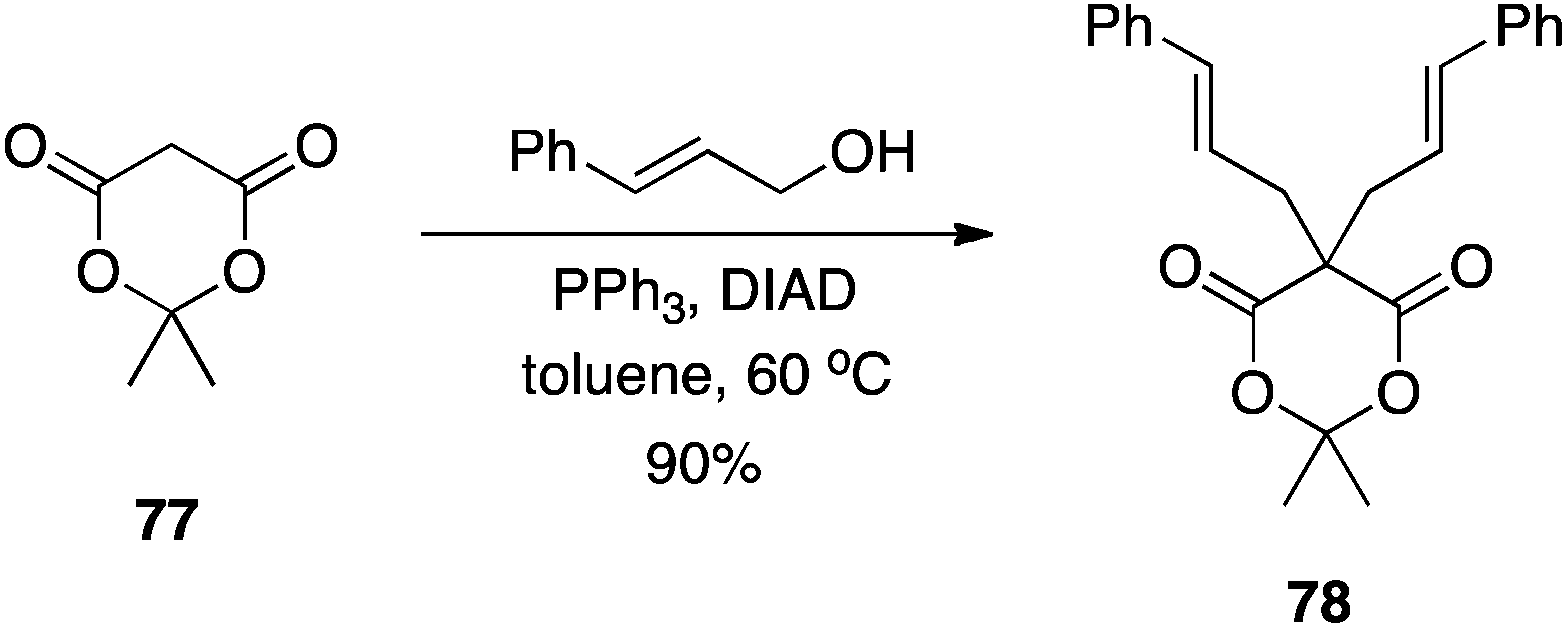
Meldrum's acid.
Meldrum's acid is a versatile synthon in organic synthesis.49 Bearing an “anomalously low” pKa of 4.97,50 it was anticipated that Meldrum's acid (77) would be an effective pronucleophile in the Mitsunobu reaction. In fact, with two acidic hydrogen atoms, it can be difficult to control the alkylation step.51 Although mono-C-alkylation of Meldrum's acid failed, Shing et al. showed that C-alkylation of mono-substituted Meldrum's acids proceeds in very good yields to furnish the 5,5-disubstituted derivatives (Scheme 23). The authors demonstrated that double C-alkylation of Meldrum's acid is also possible with moderate to very good yields reported (Scheme 24).51 Notably, 2-phenylethanol and isopropanol gave undesired with Meldrum's acid. For successful reactions to occur, the more reactive primary allylic and arylmethyl alcohols had to be employed. In reactions with secondary allylic alcohols, a catalytic amount of Pd(0) was required in order to promote the regioselectivity of C- over O-alkylation.
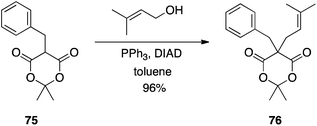 |
| | Scheme 23 C-alkylation of C5-mono-substituted Meldrum's acids. | |
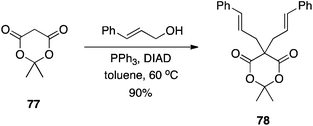 |
| | Scheme 24 Double C5-alkylation of Meldrum's acid. | |
Bis(2,2,2-trifluoroethyl) malonates.
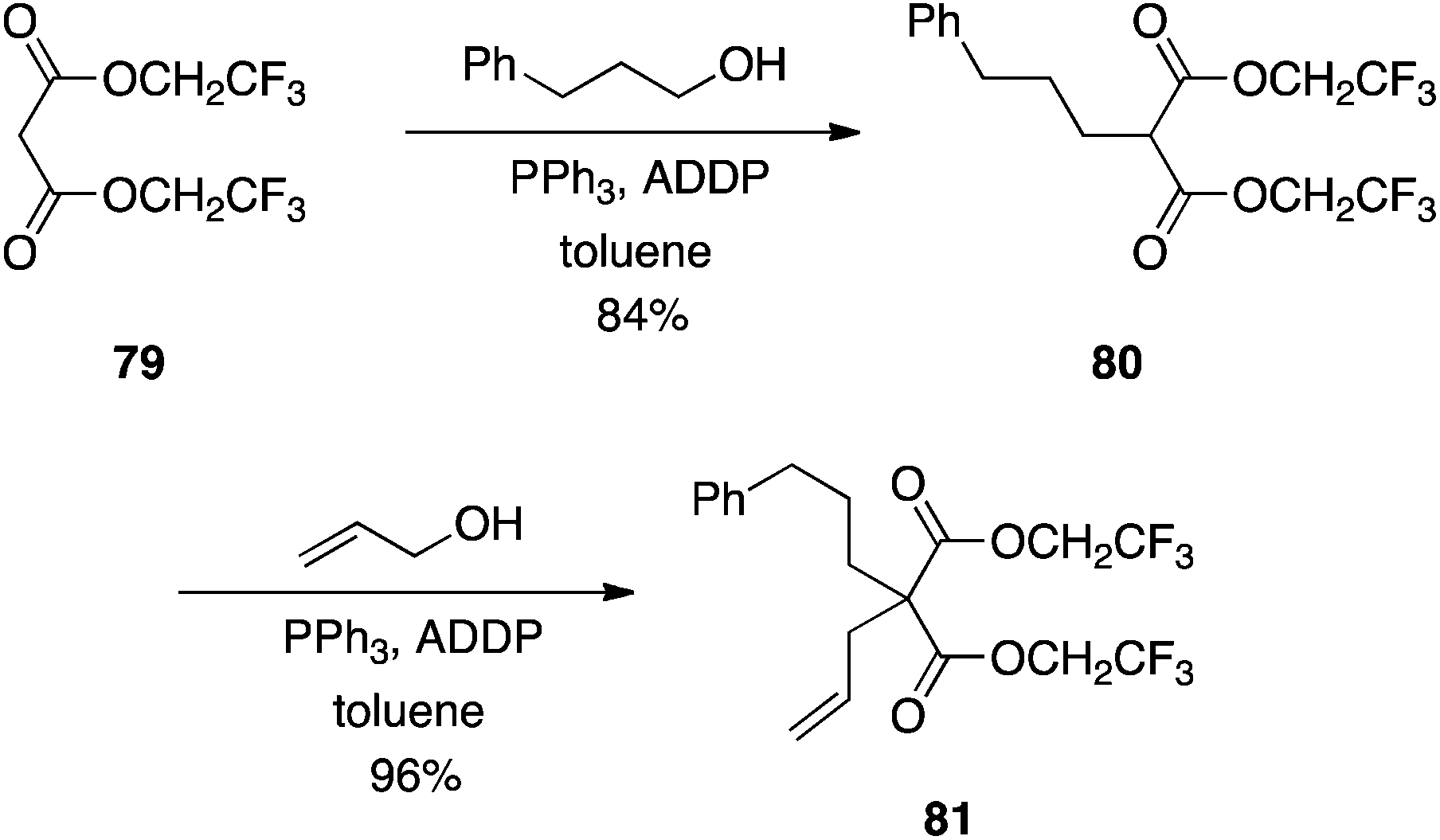
Although structurally similar to Meldrum's acid, diesters of malonic acids are poor nucleophiles in the Mitsunobu reaction46 since the activated methylene protons exhibit a much higher pKa of about 13.52 Takacs rationalized that making the alkyl components of dialkyl malonate esters more electron-withdrawing would lower the pKa of the activated methylene flanked by the two carbonyls, thereby activating it towards Mitsunobu chemistry.53 Accordingly, his group prepared the bis(2,2,2-trifluoroethyl) malonate 79, which subsequently underwent C-alkylations with primary alcohols in good to excellent yields (Scheme 25),53 with no reported O-alkylation. Secondary alcohols coupled in moderate yield at best. Importantly, however, in addition to the observed regiocontrol, Takacs found that monoalkylation could also be controlled, in contrast to analogous chemistry described with Meldrum's acid.51
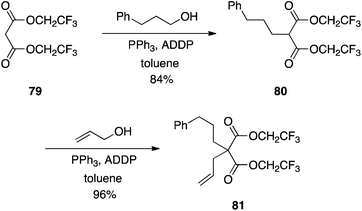 |
| | Scheme 25 Activation of the malonate methylene function with greater electron-withdrawing 2,2,2-trifluoroethyl groups allows C-alkylation to proceed smoothly. | |
Acetone cyanohydrin.
The direct conversion of primary alcohols to nitriles has been achieved with acetone cyanohydrin (83) as the HCN equivalent (Scheme 26).54 Szántay and colleagues showed that this application of the Mitsunobu reaction was compatible with secondary alcohols, too, provided they were not sterically hindered. With its avoidance of toxic HCN and metal cyanide salts, this represents a particularly attractive protocol to synthesize organic nitriles.
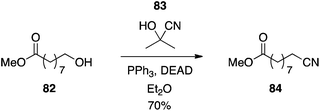 |
| | Scheme 26 Conversion of alcohols to nitriles with acetone cyanohydrin as a latent source of the pronucleophile HCN. | |
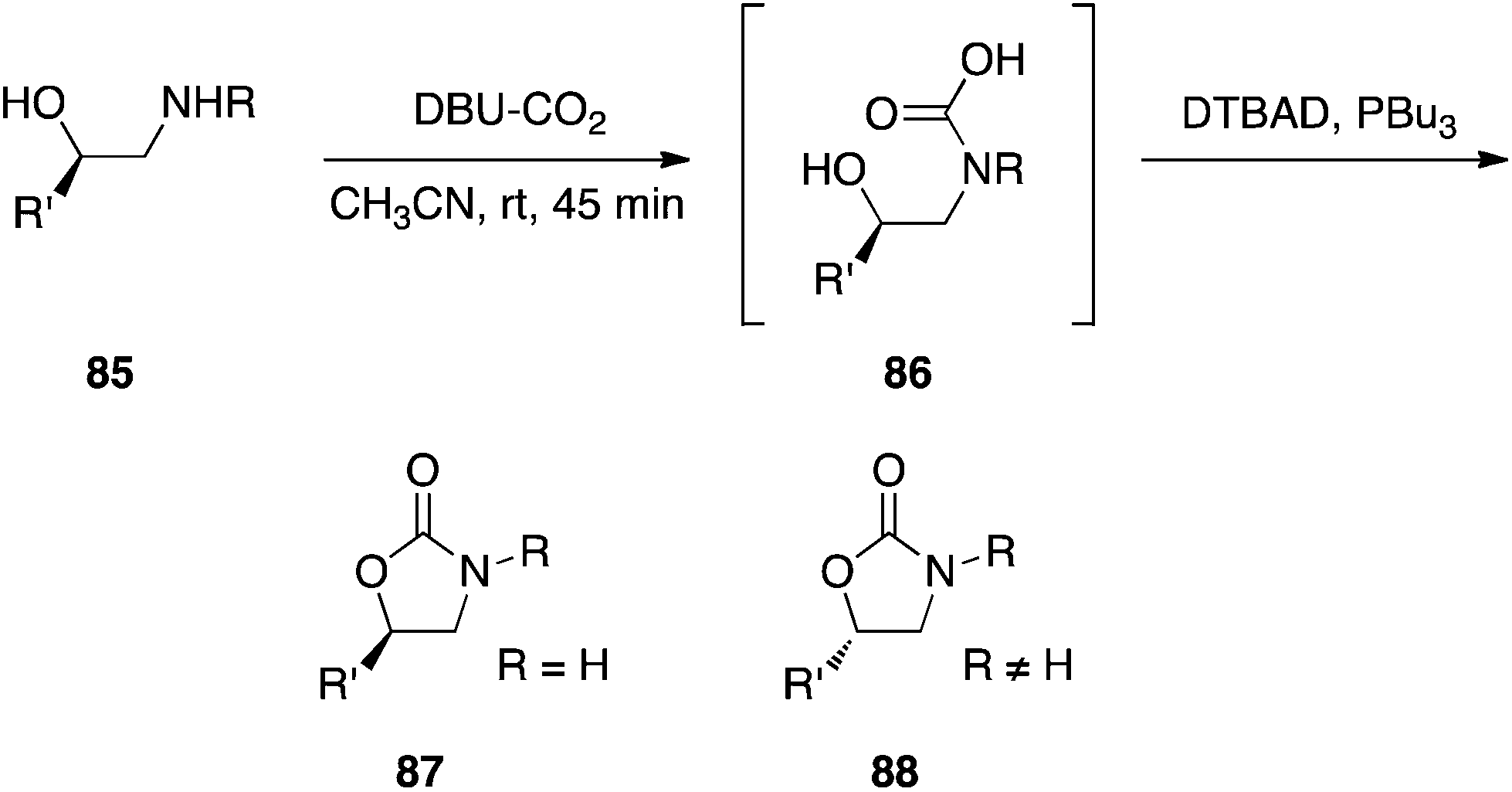
Carbamic acids.
The Mitsunobu reaction has recently been used to trap carbamic acids generated in situ as their carbamates, wherein the alcohol may be intramolecular or intermolecular.55 Interestingly, the stereochemical course of the Mitsunobu step appears to be dependent on the substitution of the nitrogen atom: when R was carbon, the expected inversion was observed; when R was hydrogen, retention was detected (Scheme 27). The authors have proposed a mechanism that accounts for this deviance from clean inversion: when R = H, the activated carbamic acid undergoes a β-elimination to afford an isocyanate that subsequently reacts with the alcohol to deliver the carbamate. Since attack does not happen at the alcohol's carbon atom, retention is observed.
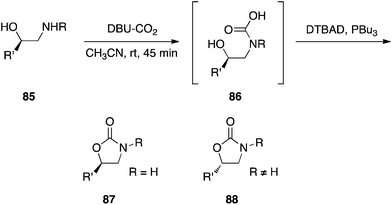 |
| | Scheme 27 Synthesis of carbamates. Note the nature of the R group dictates the stereochemical course of the reaction. | |
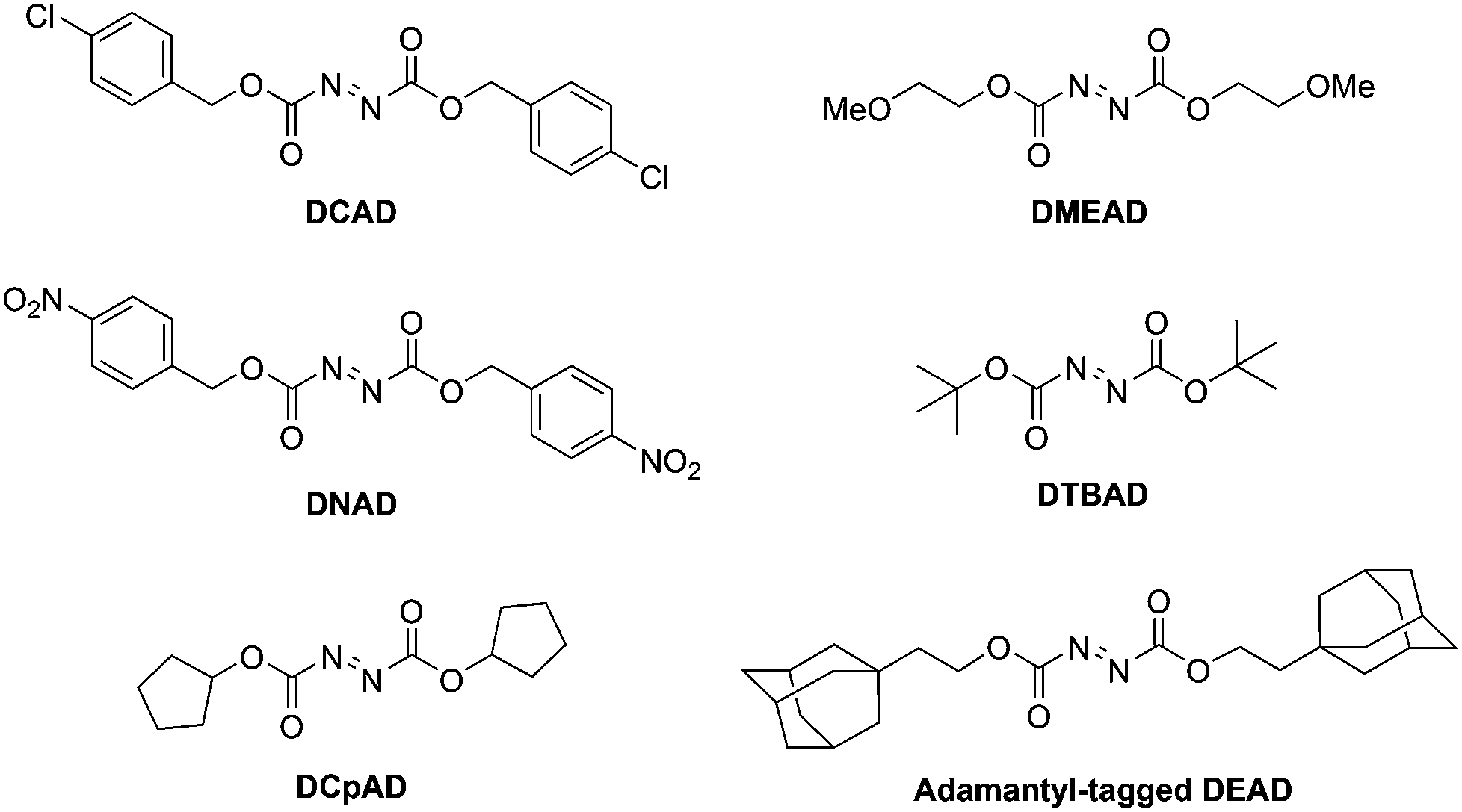
New azodicarbonyl species
DEAD and/or its hydrazine by-product DEAD-H2 (Scheme 1) can often contaminate the Mitsunobu product owing to similar Rf values.3 In the last couple of decades in particular, new DEAD analogues have emerged that facilitate purification of the reaction mixture, and many of these are presented in Scheme 28. The majority are commercially available but may, alternatively, be synthesized by reaction of hydrazine with the appropriate chloroformate, followed by oxidation to the target azo species.3
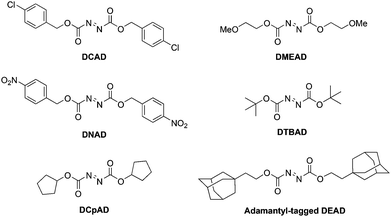 |
| | Scheme 28 DEAD/DIAD alternatives that facilitate reaction mixture purification. | |
Di-4-chlorobenzyl azodicarboxylate (DCAD).
Replacement of the ethyl groups of DEAD with 4-chlorobenzyl groups furnishes the orange solid DCAD, which is more convenient to handle than the liquids DEAD and DIAD, can be stored at room temperature and is almost as effective.56 Most of the hydrazine by-product of DCAD generated in the Mitsunobu reaction can be removed by precipitation from dichloromethane, and, owing to a different polarity to the hydrazine by-product of DIAD, purification by column chromatography may be facilitated.
Di-4-nitrobenzyl azodicarboxylate (DNAD).
Dai and colleagues recently introduced DNAD as another alternative to DEAD and DIAD.57 Like DCAD, DNAD is a crystalline solid that is stable at room temperature. DNAD is at least as efficient as DIAD but, significantly, the hydrazine by-product exhibits poor solubilities in CH2Cl2 and THF, and so can be precipitated from these solvents.
Di-cyclopentyl azodicarboxylate (DCpAD).
DCpAD represents another variant of DEAD and DIAD.58 Once again, DCpAD is an orange solid and can be stored at room temperature. DCpAD appears to be as effective as DEAD in the Mitsunobu reaction.
Di-2-methoxyethyl azodicarboxylate (DMEAD).
Chemists in Japan devised yet another analogue of DEAD in the guise of DMEAD, wherein the isopropyl groups have been replaced with 2-methoxyethyl moieties.59 DMEAD is about as effective as DIAD in Mitsunobu reactions. Importantly, though, the reduced hydrazine by-product can be removed in an aqueous work-up. Moreover, in our hands, excess DMEAD itself can also be extracted into the aqueous phase. Since Ph3PO can be removed by filtration, column chromatography may be completely avoided. DMEAD is now commercially available and, although it is somewhat more expensive than DIAD, time and money saved through evasion of chromatography render this new azodicarbonyl affordable.
Di-tert-butyl azodicarboxylate (DTBAD).
In contrast to the other DIAD variants, DTBAD's “muscle” comes from its inherent instability to acid, which results in its conversion to gaseous by-products.60 The combination of DTBAD with diphenyl(2-pyridyl)phosphine (Ph2PPy) provides an especially convenient protocol since, in addition to decomposing DTBAD (and its hydrazine by-product), work-up with acid extracts the phosphine (and its oxide) into the aqueous), indicating that column chromatography might be avoidable altogether. Like DCAD and DMEAD, DTBAD is also commercially available.
Adamantyl-tagged derivatives of DEAD.
Curran and co-workers prepared two analogues of DEAD in which the ethyl groups have been replaced with adamantly-based groups.61 The azo parents and hydrazine by-products have longer retention times on cyclodextrin-bonded silica gel than do DEAD and DEAD-H2, respectively, owing to host–guest chemistry between the adamantyl tags (guests) and the cyclodextrin (hosts). The different retention times facilitates purification of the Mitsunobu products.
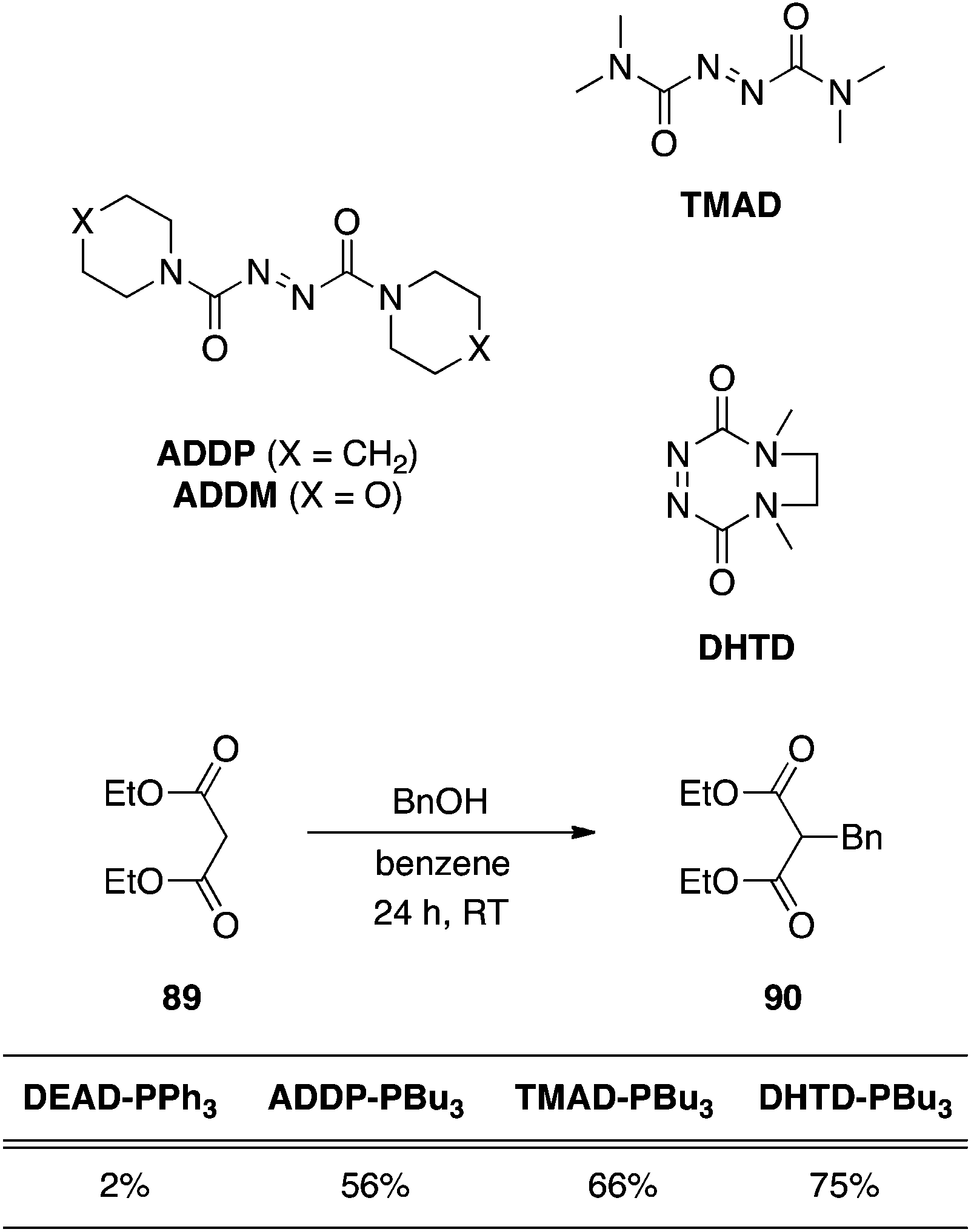
N,N,N′,N′-Tetramethyldicarboxamide (TMAD), 1,1′-(azodicarbonyl)dipiperidine (ADDP) and 4,7-dimethyl-3,5,7-hexahydro-1,2,4,7-tetrazocin-3,8-dione (DHTD).
Conversion of the esters of the azodicarboxylates to amides affords more active coupling agents;62 upon reaction with phosphines, the betaine intermediates that are generated are more basic and can deprotonate pronucleophiles exhibiting pKa values greater than 11. An impressive example of C-alkylation of diethyl malonate with 1,1′-(azodicarbonyl)dipiperidine (ADDP), N,N,N′,N′-tetramethyldicarboxamide (TMAD) and 4,7-dimethyl-3,5,7-hexahydro-1,2,4,7-tetrazocin-3,8-dione (DHTD) is shown in Scheme 29 and demonstrates their superiority over DEAD.63 An interesting note about ADDP is that it precipitates from a THF reaction medium, and can also be precipitated out by the addition of hexanes or ether.
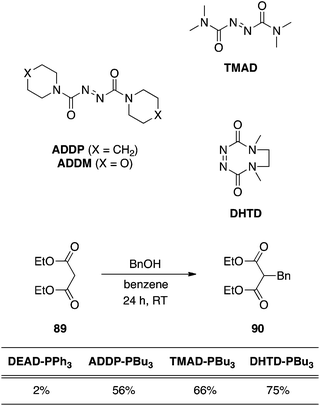 |
| | Scheme 29 Comparison of the efficiencies of Mitsunobu co-reagents in the C-alkylation of diethyl malonate. Percentages are yields of product 90. | |
Azodicarbonyl dimorpholide (ADDM).
We recently showed that the morpholine analogue of ADDP, azodicarbonyl dimorpholide (ADDM; Scheme 29), is also an effective azo species for the Mitsunobu reaction.64 An important distinction from ADDP, however, is that ADDM and its hydrazine by-product can both be completely removed from the reaction mixture by a neutral, aqueous work-up. Moreover, we demonstrated that the combination of PPh3 on resin and ADDM resulted in a chromatography-free Mitsunobu reaction.
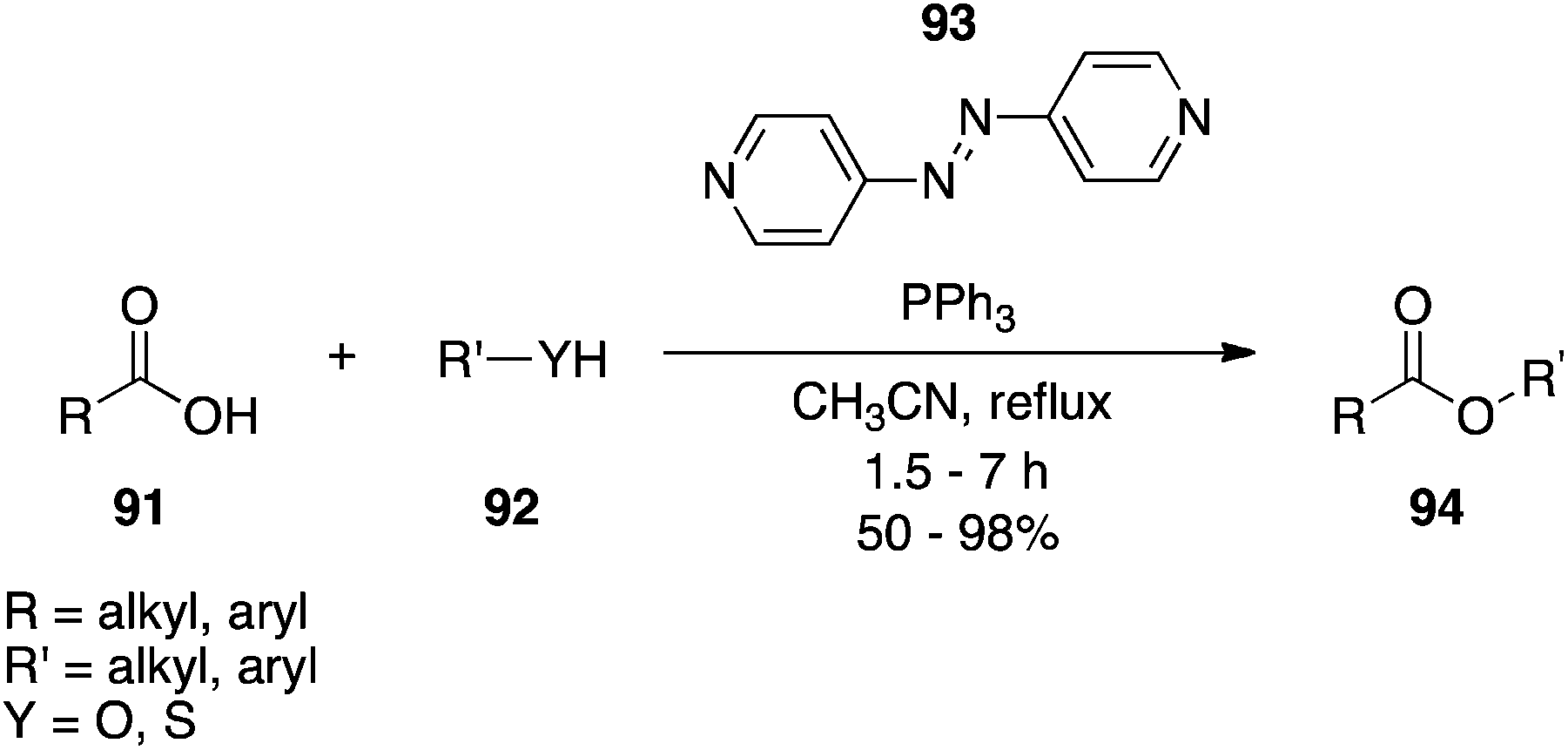
4,4′-Azopyridine.
4,4′-Azopyridine (93), which is distinct from the other DEAD surrogates owing to a lack of activating carbonyl groups, has been introduced as an alternative to DEAD (Scheme 30).65 Although reaction times are typically longer, yields are comparable. One of the main advantages of 4,4′-azopyridine over DEAD is that its by-product 4,4′-bispyridine hydrazine largely precipitates from the reaction mixture and so can mostly be removed by filtration. It is noteworthy that the hydrazine by-product can be re-oxidized to the azo form by treatment with iodosobenzene diacetate in DMSO at RT for 6 h.
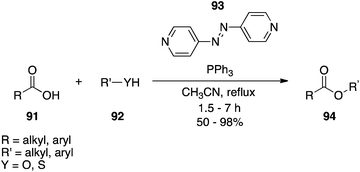 |
| | Scheme 30 4,4′-Azopyridine (93) is a suitable alternative to DIAD. | |
New phosphine species
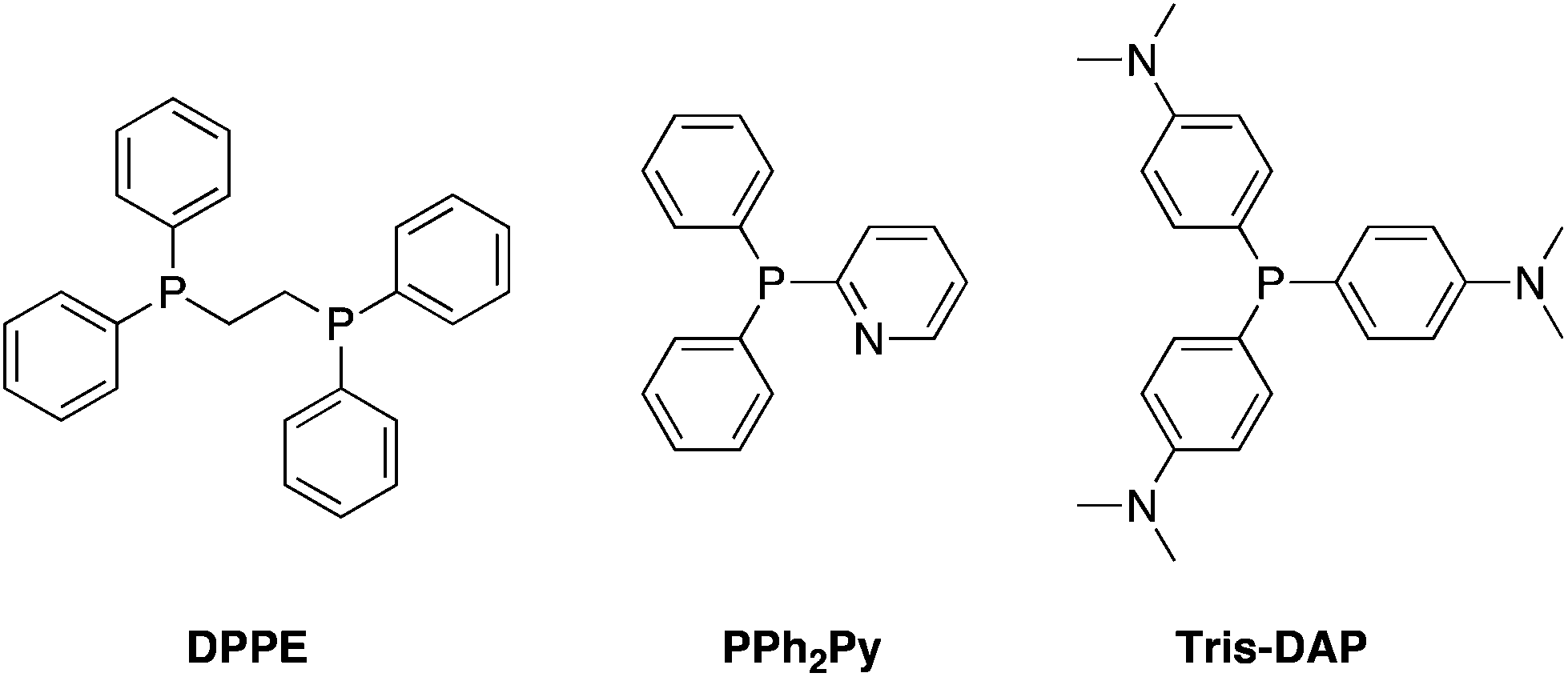
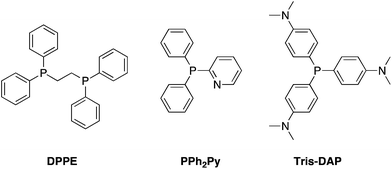
Contamination of reaction products with triphenylphosphine or triphenylphosphine oxide is arguably the biggest thorn in the Mitsunobu reaction's side.2 One of the earliest approaches to simplify the purification of the reaction mixture was to replace triphenylphosphine with tributylphosphine, since excess phosphine may be removed via evaporation and the tributylphosphine oxide is extractable into water during aqueous work-up. Although switching triphenylphosphine with tributylphosphine appears to have little effect on product yield, the latter is pyrophoric and so care should be taken during its use, especially on a large scale. 1,2-Bis(diphenylphosphino)ethane (DPPE) has also been used as a surrogate of PPh3; due to its greater polarity, the resulting bis-oxide precipitates from the reaction mixture and so can be removed by simple filtration prior to chromatography.66 Finally, in addition to multiple reports of triphenylphosphine on resin3 as a means to facilitate the purification of Mitsunobu reactions, new solution phase phosphine species have also been developed (Schemes 31–33), which will be discussed below.
 |
| | Scheme 31 PPh3 alternatives that simplify purification of the Mitsunobu reaction. | |
 |
| | Scheme 32 Phosphoranes/ylides that function as combined equivalents of phosphines and azodicarbonyls. | |
 |
| | Scheme 33 Built-in phosphine: inversion of a secondary alcohol with a carboxylic acid that also carries the requisite triarylphosphine functionality. | |
2-Pyridyl-diphenylphosphine (PPh2Py).
Replacement of one of the phenyl rings of PPh3 with a pyridine ring to give 2-pyridyl-diphenylphosphine (PPh2Py) is tolerated with no detrimental impact on the reaction yield.67 Significantly, however, the introduction of a basic nitrogen allows the removal of excess PPh2Py and its oxide by-product into an acidic aqueous layer during reaction work-up, considerably improving reaction mixture purification.
Tris(dimethylamino)phosphine (Tris-DAP).
The introduction of dimethylamino groups into the para positions of each of the phenyl rings of PPh3 affords tris(dimethylamino)phosphine, or Tris-DAP.68 Akin to Ph2PPy, Tris-DAP and its phosphine oxide by-product can also be removed by a simple acidic aqueous work-up.
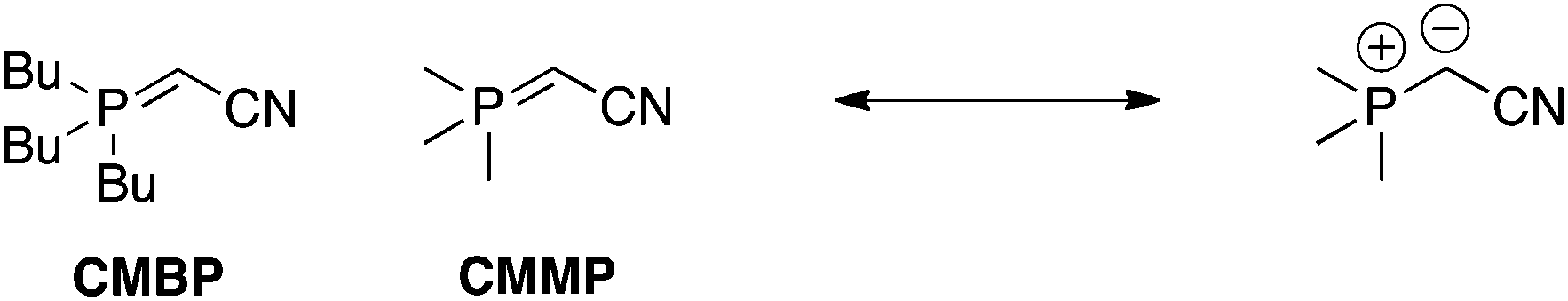
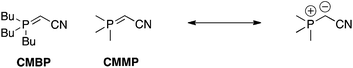
Phosphoranes/ylides.
Tsunoda and colleagues have prepared cyanomethylenephosphorane (CMMP) and cyanomethylenetributylphosphorane (CMBP) as “all-in-one” Mitsunobu reagents (Scheme 32); CMMP and CMBP serve as both the azo-type reagent and as the phosphine.69,70 The neutral phosphorane deprotonates the pronucleophile to afford a positively charged phosphonium ion, which then turns the alcohol into a good leaving group. A particular strength of these phosphoranes is that they are capable of deprotonating pronucleophiles that are normally not acidic enough for the Mitsunobu reaction. On the other hand, a couple of caveats include their limited commercial availabilities and the need for reaction temperatures in excess of the solvent boiling points, which requires re-sealable reaction vessels.
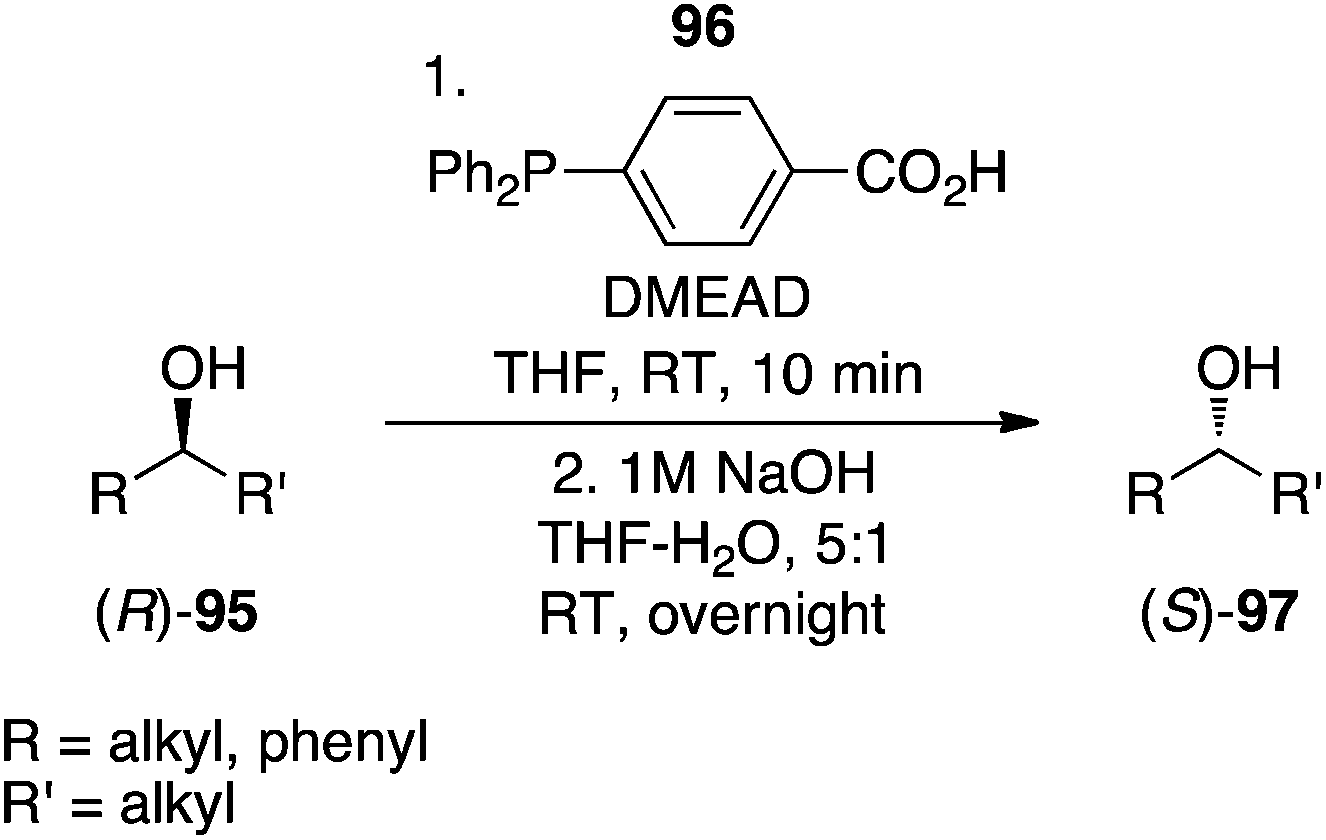
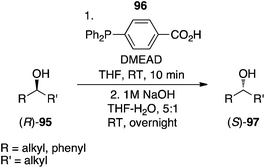
4-(Diphenylphosphino)benzoic acid.
Sugimura's group recently creatively combined a reducing agent and a pronucleophile in a single compound, 4-(diphenylphosphino)benzoic acid (96), which is commercially available.71 As illustrated in Scheme 33, secondary alcohols coupled to this phosphine in the presence of DMEAD to afford, after saponification, the corresponding inverted secondary alcohols. In addition to exhibiting high stereochemical purities, the compounds were sufficiently chemically pure owing to the extractability of DMEAD/DMEAD-H2 and 4-(diphenylphosphoryl)benzoic acid into the aqueous layer during work-up.
Organocatalytic Mitsunobu reactions
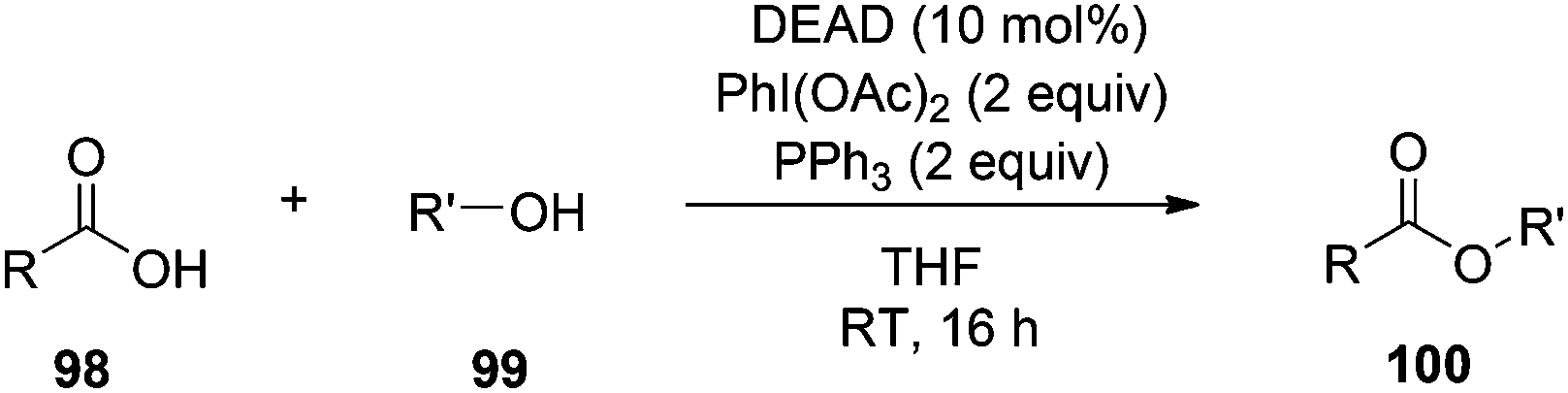
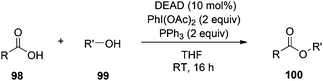
One of the drawbacks of the Mitsunobu reaction is poor atom economy. The “Holy Grail” of Mitsunobu chemistry would be to require only catalytic quantities of the azodicarbonyl and phosphine species. Towards this end, Toy and co-workers have introduced a modification to the procedure that requires only 10 mol% of DEAD whose hydrazine by-product is constantly re-oxidized in situ by the additive PhI(OAc)2 (Scheme 34).72 However, 2 equiv of PhI(OAc)2 are required (as well as 2 equiv of PPh3), negating the reduction of DEAD in terms of atom economy (moreover, prices of PhI(OAc)2 and DEAD are comparable), and acetic acid is generated during the re-oxidation of DEAD-H2, which could inadvertently participate in the Mitsunobu reaction as the pronucleophile. Nevertheless, this represents an exciting piece of work in Mitsunobu history.
 |
| | Scheme 34 Towards an organocatalytic Mitsunobu reaction. Note the sub-stoichiometric use of DIAD. | |
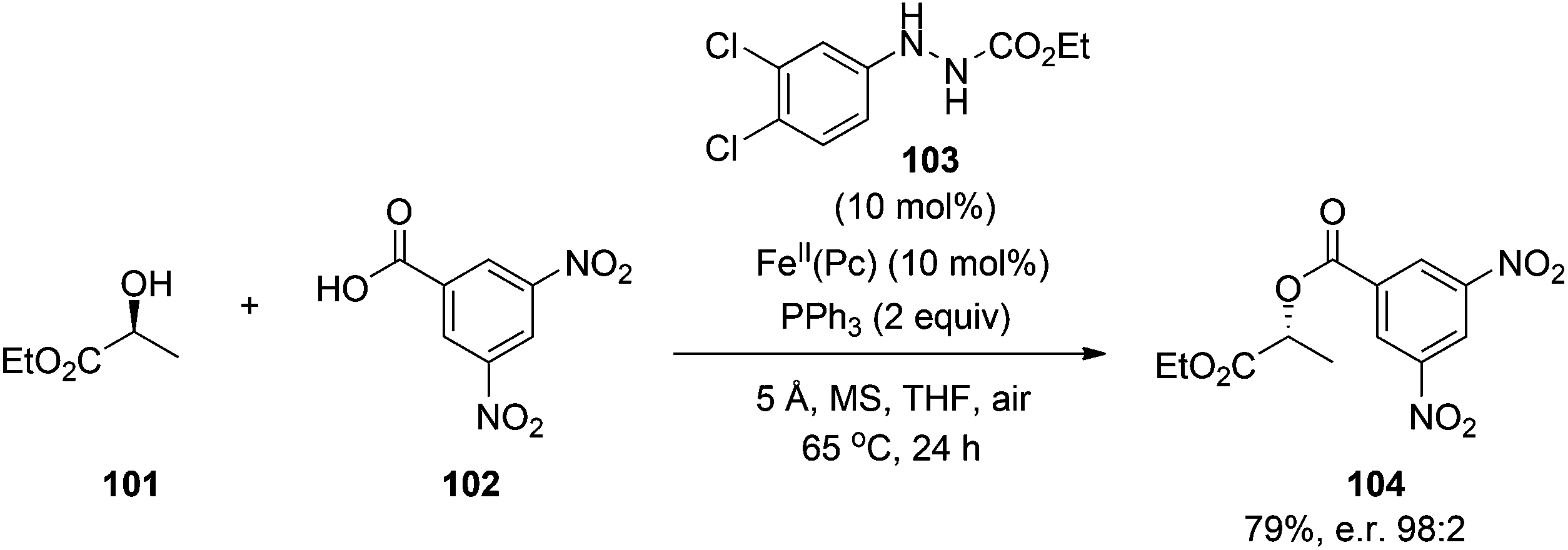
Ishibashi and colleagues73 have also made progress towards a catalytic Mitsunobu reaction. Inspired by their findings that iron phthalocyanine (FeII(Pc)) catalyzes the aerobic oxidation of carbazates (RCO2NHNH2), they envisaged that FeII(Pc) might also catalyze the aerobic re-oxidation of DEAD-H2 back to DEAD. Although attempts to accomplish this goal with 10 mol% FeII(Pc) were unsuccessful, replacement of one of the esters of DEAD-H2 with a phenyl group (i.e. PhNHNHCO2Et) proved fruitful. After screening solvents, additives, concentration and temperature, it was found that activated molecular sieves (MS) were also required to ensure that the Mitsunobu reaction proceeded, albeit in only 17% yield, with catalytic DEAD-H2 and FeII(Pc) as a two-component substitute for stoichiometric DEAD. Towards the optimization of this catalytic Mitsunobu reaction, the group prepared a series of 14 hydrazine catalysts, ArNHNHCO2Et, and discovered that hydrazine 103 was the most effective for the coupling in Scheme 35, both in terms of product yield (79%) and stereoselectivity, with almost complete inversion observed (98:2). Their improved reaction conditions were also successfully applied to a variety of alcohols and pronucleophiles, including an imide and a sulfonamide. Whilst yields are presently considerably lower than reactions with stoichiometric reagents, this is an impressive advancement in the development of a catalytic Mitsunobu reaction. In contrast to Toy's work, there are no (pro)nucleophiles in the additive reagent that might inadvertently participate in the reaction suggesting that Ishibashi's strategy has significant potential. Further optimization of the reaction conditions should focus on the reduction of the phosphine reagent to catalytic quantities, providing a truly catalytic Mitsunobu reaction.
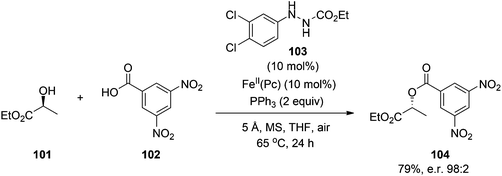 |
| | Scheme 35 A catalytic Mitsunobu reaction employing an iron catalyst and air as a terminal oxidizer. | |
Other strategies to facilitate purification
Fluorous Mitsunobu reaction.
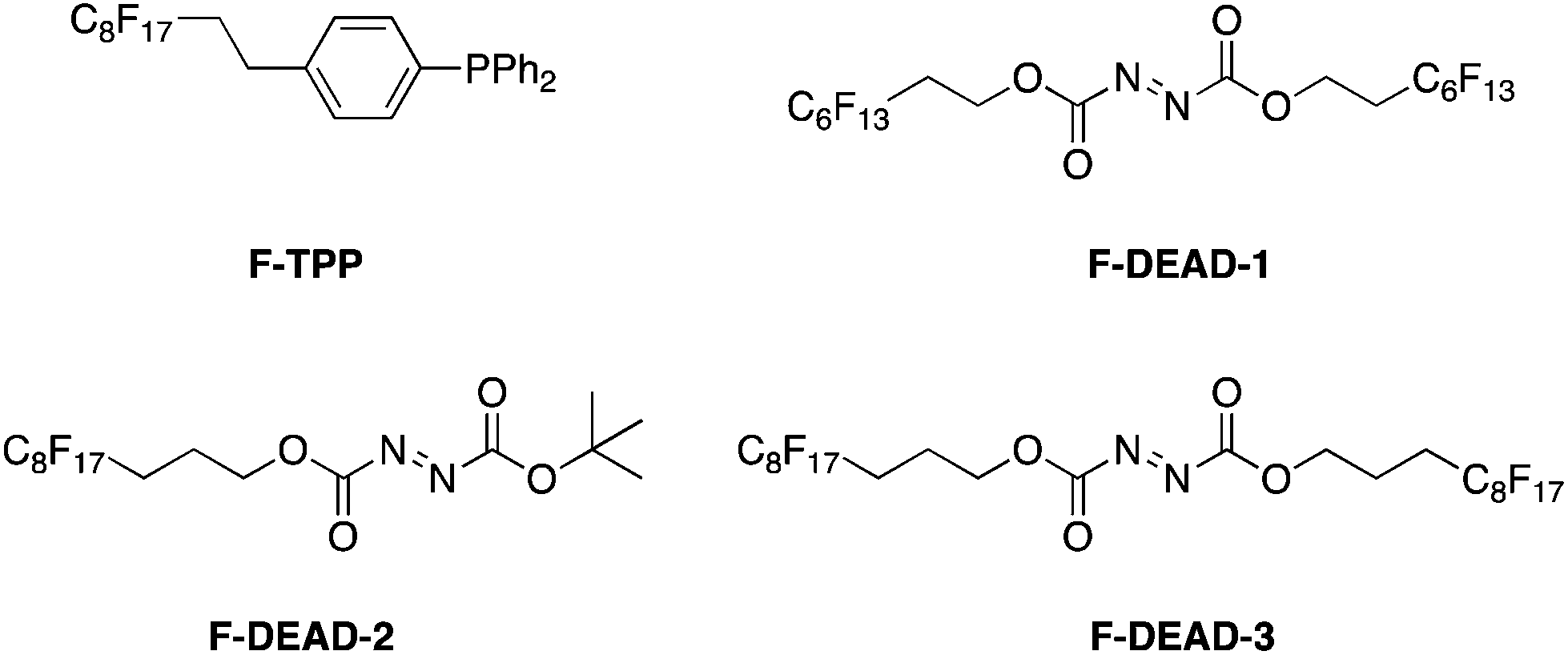
Curran introduced fluorous reverse phase silica gel at the turn of this Century, and demonstrated that materials partition over the silica at different rates according to their fluorine content.74 His group later applied this as a means to simplify purification of Mitsunobu reactions.75 Although F-DEAD-1 and F-TPP, the fluorous analogues of DEAD and PPh3, respectively (Scheme 36), successfully promoted the coupling of alcohols to acidic pronucleophiles, yields were typically very low. Whilst this shortcoming may be considered offset by the easier purification of the reaction mixture through fluorous solid-phase extraction (FSPE) relative to a non-fluorous Mitsunobu reaction, Curran and colleagues sought to improve the efficiency of their fluorous reagents. Accordingly, a library of second generation fluorous azodicarboxylates was prepared.76 Pleasingly, F-DEAD-2 and F-DEAD-3 demonstrated greatly improved reactivities, which has been attributed to the propyl spacer in place of an ethyl spacer that renders the resulting betaine sufficiently basic to deprotonate the pronucleophile.
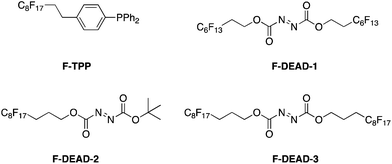 |
| | Scheme 36 Fluorous Mitsunobu reagents to facilitate purification of the reaction mixture. | |
Hydrogen peroxide.
The use of a 15 wt% solution of hydrogen peroxide (H2O2) has been described to oxidize all remaining PPh3 to Ph3PO.77 Interestingly, the hydrazine by-product of DIAD did not get oxidized back to DIAD. Regardless, this exposure to H2O2 significantly facilitated purification with the requirement of column chromatography simply over a plug of silica gel. It is anticipated that reactions performed on a large scale will particularly benefit from this purification strategy.
Miscellaneous
Sonication promotes the Mitsunobu reaction of phenols with sterically encumbered alcohols.
He and Lepore have demonstrated that sonication affords dramatic rate enhancements in the coupling of alcohols to phenols in which either or both reaction partners is/are sterically hindered (Scheme 37).78 The authors noted that the increased rate of reaction might be related to the generation of free radicals, which suggests there may be an erosion of stereospecificity in the Mitsunobu reaction; this was not explored further. Also, the reader is cautioned against performing this reaction on a multi-gram scale, as the heat generated by sonication might cause the azodicarboxylate species to explode. Nevertheless, the chemistry appears useful for the milligram scale.
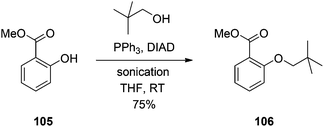 |
| | Scheme 37 Sonication assists in difficult Mitsunobu etherifications. | |
Reductive deoxygenation.
The traditional approach to accomplish the reductive deoxygenation of alcohols involves first the acylation of the alcohol with thiocarbonyldiimidazole followed by reductive cleavage mediated by tri-n-butyltin hydride.79 Myers and colleagues have introduced a single-step procedure to achieve the same goal.79 Particularly, conducting the Mitsunobu reaction with the pronucleophile O-nitrobenzenesulfonylhydrazine (44) at −30 °C yields the corresponding 1-alkyl-1-sulfonylhydrazine. Warming to 0 °C results in gentle extrusion of o-nitrobenzenesulfinic acid and the concomitant formation of a diazene intermediate that slowly loses N2 gas to furnish the deoxygenated product (Scheme 38). The chemistry appears to occur through a free-radical mechanism and works well for sterically unencumbered substrates.80
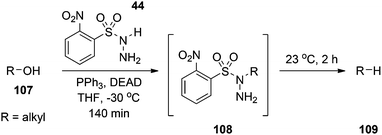 |
| | Scheme 38 Deoxygenation of alcohols to alkanes using o-nitrobenzenesulfonylhydrazine 44. | |
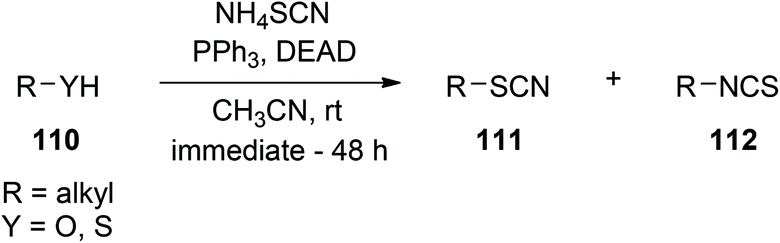
Synthesis of thiocyanates (and isothiocyanates).
Introduction of NH4SCN into the Mitsunobu protocol permits the conversion of alcohols, thiols, carboxylic acids, as well as TMS ethers and esters, to their corresponding thiocyanates and isothiocyanates, as depicted in Scheme 39. Yields are good, although 2 eq. of DEAD and PPh3 are required to consume all of the starting materials.81
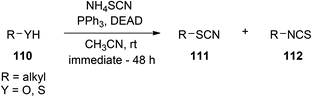 |
| | Scheme 39 Synthesis of thiocyanates and isothiocyanates with the thiocyanate anion as the nucleophile. | |
Conclusions
The Mitsunobu reaction between an alcohol and a pronucleophile has become a standard chemical reaction in many laboratories, which is largely due to its mildness, occurring essentially under neutral conditions, its speed, many reactions are often complete within 1 h, and clean inversion of the alcohol's stereocentre. The scope has been expanded beyond the traditional pronucleophiles to encompass oximes, purines and β-ketoesters to name but a few, permitting the construction of C–O, C–N and C–C bonds, respectively. At the same time as the expansion of the pronucleophile cache, research into new reagents to facilitate purification of the reaction mixture has been rife. In particular, new azodicarbonyls, such as DMEAD and ADDM, may be extracted into water during a standard work-up, whilst PPh2Py may be extracted into the acidic aqueous. Some progress has also been made towards a catalytic Mitsunobu reaction, widely regarded as the “Holy Grail” of Mitsunobu chemistry. We hope that further advancements are made in this regard as it would address two caveats of the Mitsunobu reaction: problematic purification and poor atom economy. Finally, an area of Mitsunobu chemistry that would benefit greatly from further research is C–C bond formation, as this has been somewhat neglected.
Acknowledgements
The University of Maryland School of Pharmacy is acknowledged for supporting our work in this area.
Notes and references
- O. Mitsunobu and M. Yamada, Bull. Chem. Soc. Jpn., 1967, 40, 2380 CrossRef CAS.
- O. Mitsunobu, Synthesis, 1981, 1 CrossRef CAS.
- K. C. Kumara Swamy, N. N. Bhuvan Kumar, E. Balaraman and K. V. P. Pavan Kumar, Chem. Rev., 2009, 109, 2551 CrossRef PubMed.
- D. L. Hughes, Org. Prep., 1996, 28, 127 CrossRef CAS.
- T. Y. S. But and P. H. Toy, Chem. – Asian J., 2007, 2, 1340 CrossRef CAS PubMed.
- A. V. Stachulski, J. Chem. Soc., Perkin Trans. 1, 1991, 3065 RSC.
- J. W. Westly, J. W. Liu, J. F. Blount, L. H. Sello, N. Troupe and P. A. Miller, J. Antibiot., 1983, 36, 1275 CrossRef CAS; R. D. Hsugwitz, R. G. Angel, G. A. Jacobs, B. V. Maurer, V. L. Narayanan, L. R. Cruthers and J. Szanto, J. Med. Chem., 1982, 25, 969 CrossRef; Y. Katsura, S. Nishino, Y. Inoue, M. Tomoi and H. Tskasugi, Chem. Pharm. Bull., 1992, 40, 371 CrossRef.
- F. Wang and J. R. Hauske, Tetrahedron Lett., 1997, 38, 6529 CrossRef CAS.
- Y. Yan, Q. Zhong, N. Zhao and G. Liu, Mol. Diversity, 2012, 16, 157 CrossRef CAS PubMed.
- D. Röthlisberger, O. Khersonsky, A. M. Wollacott, L. Jiang, J. DeChancie, J. Betker, J. L. Gallaher, E. A. Althoff, A. Zanghellini, O. Dym, S. Albeck, K. N. Houk, D. S. Tawfik and D. Baker, Nature, 2008, 453, 190 CrossRef PubMed.
- M. L. Casey, D. S. Kemp, K. G. Paul and D. D. Cox, J. Org. Chem., 1973, 38, 2294 CrossRef CAS.
- G. Poissonnet, Synth. Commun., 1997, 27, 3839 CrossRef CAS.
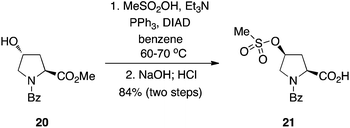
- L. Chen, M. E. Lanning and S. Fletcher, Aust. J. Anal. Pharm. Chem., 2014, 1, 1015 Search PubMed.
- C. Ballatore, D. M. Huryn and A. B. Smith 3rd, ChemMedChem, 2013, 8, 385 CrossRef CAS PubMed.
-
CRC Handbook of Chemistry and Physics, ed. W. M. Haynes, CRC Press, Boca Raton, FL, 95th edn, 2014–2015 Search PubMed.
- E. Whiting, M. E. Lanning, J. A. Scheenstra and S. Fletcher, J. Org. Chem., 2015, 80, 1229 CrossRef CAS PubMed.
- N. G. Anderson, D. A. Lust, K. A. Colapret, J. H. Simpson, M. F. Malley and J. Z. Gougoutas, J. Org. Chem., 1996, 60, 7955 CrossRef.
- L. Matzen, A. Engesgaard, B. Ebert, M. Didriksen, B. Frølund, P. Krogsgaard-Larsen and J. W. Jaroszewski, J. Med. Chem., 1997, 40, 520 CrossRef CAS PubMed.
- B. Frolund, A. T. Jorgensen, L. Tagmose, T. B. Stensbol, H. T. Vestergaard, C. Engblom, U. Kristiansen, C. Sanchez, P. Krogsgaard-Larsen and T. Liljefors, J. Med. Chem., 2002, 45, 2454 CrossRef CAS PubMed.
- G. Schlewer and P. Krogsgaard-Larsen, Acta Chem. Scand. Ser. B, 1984, 38, 815 CrossRef PubMed.
- L. Chen and S. Fletcher, Tetrahedron Lett., 2014, 55, 1693 CrossRef CAS PubMed.
- C. Simon, S. Hosztafi and S. Makleit, Tetrahedron, 1994, 50, 9757 CrossRef CAS.
- J. R. Henry, L. R. Marcin, M. C. McIntosh, R. M. Scola, G. D. Harris Jr. and S. M. Weinreb, Tetrahedron Lett., 1989, 30, 5709 CrossRef CAS.
- T. Fukuyama, C.-K. Jow and M. Cheung, Tetrahedron Lett., 1995, 36, 6373 CrossRef CAS.
- T. Kan and T. Fukuyama, Chem. Commun., 2004, 353 RSC.
- S. Kobayashi, G. Peng and T. Fukuyama, Tetrahedron Lett., 1999, 40, 1519 CrossRef CAS.
- X. Cai, M. S. Chorghade, A. Fura, G. S. Grewal, K. A. Jauregui, H. A. Lounsbury, R. T. Scannell, C. G. Yeh, M. A. Young and S. Yu, Org. Proc. R&D, 1999, 3, 73 CAS.
- J. E. Green, D. M. Bender, S. Jackson, M. J. O'Donnell and J. R. McCarthy, Org. Lett., 2009, 11, 807 CrossRef CAS PubMed.
- H.-O. Kim and M. Kahn, Synlett, 1999, 1239 CrossRef CAS PubMed.
- A. G. Myers and B. Zheng, J. Am. Chem. Soc., 1996, 118, 4492 CrossRef CAS.
- K. S. Dodd and A. P. Kozikowski, Tetrahedron Lett., 1994, 35, 977 CrossRef.
- H.-O. Kim, F. Matthew and C. Ogbu, Synlett, 1999, 193 CrossRef CAS PubMed.
- Y. Kitade, T. Ando, T. Yamaguchi, A. Hori, M. Nakanishi and Y. Ueno, Bioorg. Med. Chem., 2006, 14, 5578 CrossRef CAS PubMed; M. Yang, W. Ye and S. W. Schneller, J. Org. Chem., 2004, 69, 3993 CrossRef PubMed.
- W. Lu, S. Sengupta, J. L. Petersen, N. G. Akhmedov and X. Shi, J. Org. Chem., 2007, 72, 5012 CrossRef CAS PubMed.
- E. A. Véliz and P. A. Beal, Tetrahedron Lett., 2006, 47, 3153 CrossRef PubMed.
- J. Kjellberg and N. G. Johansson, Tetrahedron, 1986, 42, 6541 CrossRef CAS.
- S. Fletcher, V. M. Shahani and P. T. Gunning, Tetrahedron Lett., 2009, 50, 4258 CrossRef CAS PubMed.
- S. Fletcher, V. M. Shahani, A. J. Lough and P. T. Gunning, Tetrahedron, 2010, 66, 4621 CrossRef CAS PubMed.
- S. Fletcher, Tetrahedron Lett., 2010, 51, 2948 CrossRef CAS PubMed.
- W. C. Ripka, G. V. De Lucca, A. C. Bach II, R. S. Pottorf and J. M. Blaney, Tetrahedron, 1993, 49, 3593 CrossRef CAS.
- M. D. Cummings, C. Schubert, D. J. Parks, R. R. Calvo, L. V. LaFrance, J. Lattanze, K. L. Milkiewicz and T. Lu, Chem. Biol. Drug Des., 2006, 67, 201 CAS.
- B. E. Blass, T. M. Burt, S. Liu, D. E. Portlock and E. M. Swing, Tetrahedron Lett., 2000, 41, 2063 CrossRef CAS.
- K.-Y. Jung and S. Fletcher, Med. Chem. Commun., 2012, 3, 1160 RSC.
- J. M. Keith and L. Gomez, J. Org. Chem., 2006, 71, 7113 CrossRef CAS PubMed.
-
E. J. Corey and X.-M. Cheng, The Logic of Chemical Synthesis, Wiley, 1995 Search PubMed.
- M. Wada and O. Mitsunobu, Tetrahedron Lett., 1972, 13, 1279 CrossRef.
- S. Manna and J. R. Falck, Synth. Commun., 1985, 15, 663 CrossRef CAS.
- G. Cravotto, G. B. Giovenzana, M. Sisti and G. Palmisano, Tetrahedron, 1996, 52, 13007 CrossRef CAS.
- Y. Oikawa, H. Hirasawa and O. Yonemitsu, Tetrahedron Lett., 1978, 19, 1759 CrossRef.
- S. Nakamura, H. Hirao and T. Ohwada, J. Org. Chem., 2004, 69, 4309 CrossRef CAS PubMed.
- T. K. M. Shing, L.-H. Li and K. Narkunan, J. Org. Chem., 1997, 62, 1617 CrossRef CAS.
- R. G. Pearson and R. L. Dillon, J. Am. Chem. Soc., 1953, 75, 2439 CrossRef CAS.
- J. M. Takacs, Z. Xu, X.-T. Jiang, A. P. Leonov and G. C. Theriot, Org. Lett., 2002, 4, 3843 CrossRef CAS.
- M. C. Aesa, G. Baán, L. Novák and C. Szántay, Synth. Commun., 1996, 26, 909 CrossRef CAS.
- C. J. Dinsmore and S. P. Mercer, Org. Lett., 2004, 6, 2885 CrossRef CAS PubMed.
- B. H. Lipshutz, D. W. Chung, B. Rich and R. Corral, Org. Lett., 2006, 8, 5069 CrossRef CAS PubMed.
- J. Yang, L. Dai, X. Wang and Y. Chen, Tetrahedron, 2011, 67, 1456 CrossRef CAS PubMed.
- J. Yang, L. Dai, X. Z. Wang and Y. Chen, Chin. Chem. Lett., 2011, 22, 1047 CrossRef CAS PubMed.
- K. Hagiya, N. Muramoto, T. Misaki and T. Sugimura, Tetrahedron, 2009, 65, 6109 CrossRef CAS PubMed.
- M. Kiankarimi, R. Lowe, J. R. McCarthy and J. P. Whitten, Tetrahedron Lett., 1999, 40, 4497 CrossRef CAS.
- S. Dandapani, J. J. Newsome and D. P. Curran, Tetrahedron Lett., 2004, 45, 6653 CrossRef CAS PubMed.
- T. Tsunoda, Y. Yamamiya and S. Itô, Tetrahedron Lett., 1993, 34, 1639 CrossRef CAS.
- T. Tsunoda, M. Nagaku, C. Nagino, Y. Kawamura, F. Ozaki, H. Hioki and S. Itô, Tetrahedron Lett., 1995, 36, 2531 CrossRef CAS.
- M. E. Lanning and S. Fletcher, Tetrahedron Lett., 2013, 54, 4624 CrossRef CAS PubMed.
- N. Iranpoor, H. Firouzabadi, D. Khalili and S. Motevalli, J. Org. Chem., 2008, 73, 4882 CrossRef CAS PubMed.
- I. A. O'Neil, S. Thompson, C. L. Murray and S. B. Kalindjian, Tetrahedron Lett., 1998, 39, 7787 CrossRef.
- D. Camp and I. D. Jenkina, Aust. J. Chem., 1988, 41, 1835 CrossRef CAS.
- G. Grynkiewicz, J. Jurcazk and A. Zamojski, Tetrahedron, 1975, 31, 1411 CrossRef CAS.
- T. Tsunoda, F. Ozaki and S. Itô, Tetrahedron Lett., 1994, 35, 5081 CrossRef CAS.
- T. Tsunoda, C. Nagino, M. Oguri and S. Itô, Tetrahedron Lett., 1996, 37, 2459 CrossRef CAS.
- N. Muramoto, K. Yoshino, T. Misaki and T. Sugimura, Synthesis, 2013, 931 CAS.
- T. Y. But and P. H. Toy, J. Am. Chem. Soc., 2006, 128, 9636 CrossRef CAS PubMed.
- D. Hirose, T. Taniguchi and H. Ishibashi, Angew. Chem. Int. Ed., 2013, 52, 4613 CrossRef CAS PubMed.
-
D. P. Curran, S. Hadida, A. Studer, M. He, S.-Y. Kim, Z. Luo, M. Larhed, M. Hallberg and B. Linclau, Fluorous Synthesis: A User's Guide, in Combinatorial Chemistry: A Practical Approach, ed. H. Fenniri, Oxford Univ Press, Oxford, 2000, vol. 2, p. 327 Search PubMed.
- S. Dandapani and D. P. Curran, Tetrahedron, 2002, 58, 3855 CrossRef CAS.
- S. Dandapani and D. P. Curran, J. Org. Chem., 2004, 69, 8751 CrossRef CAS PubMed.
- A. J. Proctor, K. Beautement, J. M. Clough, D. W. Knight and Y. Li, Tetrahedron Lett., 2006, 47, 5151 CrossRef CAS PubMed.
- S. D. Lepore and Y. He, J. Org. Chem., 2003, 68, 8261 CrossRef CAS PubMed.
- D. H. R. Barton and S. W. McCombie, J. Chem. Soc., Perkin Trans. 1, 1975, 1574 RSC.
- A. G. Myers, M. Movassaghi and B. Zheng, J. Am. Chem. Soc., 1997, 119, 8572 CrossRef CAS.
- N. Iranpoor, H. Firouzabadi, B. Akhlaghinia and R. Azadi, Synthesis, 2004, 92 CrossRef CAS PubMed.
|
| This journal is © the Partner Organisations 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.