
The Mitsunobu reaction in the 21st century
Abstract
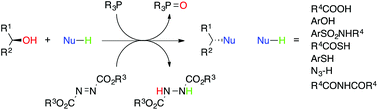
The Mitsunobu reaction was first described almost fifty years ago and has enjoyed immense popularity since its inception. The purpose of this review is to focus on the more recent advances and applications of Mitsunobu chemistry, particularly from the 1990s to the present day. In addition to a discussion of newer reagents that facilitate purification, we will describe more contemporary applications of this chemistry, especially as applied to the synthesis of pharmaceuticals and their precursors.
- This article is part of the themed collection: Organic Chemistry Frontiers Outstanding Paper Awards 2014-2023
 Please wait while we load your content...
Please wait while we load your content...

