
Simple, versatile, and chemoselective reduction of secondary amides and lactams to amines with the Tf2O–NaBH4 or Cp2ZrHCl–NaBH4 system†
Pei-Qiang
Huang
*ab and
Hui
Geng
a
aDepartment of Chemistry and Fujian Provincial Key Laboratory of Chemical Biology, Collaborative Innovation Centre of Chemistry for Energy Materials, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, Fujian 361005, PR China. E-mail: pqhuang@xmu.edu.cn
bState Key Laboratory of Applied Organic Chemistry Lanzhou University, Lanzhou 730000, China
First published on 6th January 2015
Abstract
The reduction of secondary amides to amines is an important transformation for the total synthesis of alkaloids and pharmaceuticals. General and chemoselective direct methods for this transformation are scarce. We report in this paper a simple method for the direct reduction of secondary amides, which uses only two reagents, triflic anhydride for amide activation, and NaBH4 for reduction. Running under mild conditions (0 °C to r.t.), the reaction works well with several types of secondary amides including aromatic amides, aliphatic amides, and α,β-unsaturated amides. The reaction displays a good functional group tolerance for a series of reducible functional groups. The method is applicable to the direct reduction of C–H lithiation-functionalization products, and C–H activation-functionalization products. Moreover, chemoselective reduction of secondary lactams has been achieved using Cp2ZrHCl–NaBH4 combination.
Introduction
The reduction of amides to amines is an important transformation for the synthesis of bioactive alkaloids and N-containing medicinal agents.1 Due to the high stability of amides, use of strong reducing agents2 such as LiAlH4,3 diborane,4 borane-THF/SMe2 complex,5 and DIBAL-H6 is generally required. In addition, sometimes the reduction reaction needs to be run under harsh conditions. As for the reduction of secondary amides, the classical Brown reduction with the borane-THF/SMe2 complex is incompatible with alkenyl and alkynyl groups, and requires an extra step to dissociate borane–amine adducts.7 It is not surprising that the classical methods have low functional group tolerance and may lead to undesired products.3,8 These factors can account for the fact that stepwise protocols involving the pre-transformation of amides to thioamides1e–i,l or the pre-transformation of secondary amides to imides9 are methods of choice in the synthesis of alkaloids and medicinal agents.1 To suit the needs of modern organic synthesis and pharmaceutical chemistry, development of chemoselective methods for the direct reduction of amides has attracted considerable attention, and significant progress has been made in recent years.10,11 However, chemoselective reduction of amides under mild conditions remains a challenging issue,10a,b,e in particular for secondary amides,12 which contain both a highly stabilized carbonyl and an active proton. Only limited examples involving chemoselective reduction of secondary amides have been reported.12General and chemoselective methods with a broad functional group tolerance are rare, which include Charette's chemoselective reduction utilizing the Tf2O/2-F-Py/Et3SiH/Hantzsch ester hydride (HEH) system,12b Nagashima's Ru11e and Pt10l catalyzed hydrosilylations, Beller's zinc-catalyzed chemoselective reduction,12c and boronic acid catalyzed selective hydrosilylation,12i Reeves’ Ru-catalyzed hydrosilylation,12f as well as Luo's low-valent titanium reduction.12g Moreover, secondary amides serve as powerful directing groups for both directed lithiation-functionalization13 and C–H activation-functionalization.14 Thus, simple methods for direct and chemoselective reduction of secondary amides are still highly demanding.15
As a continuation of our program on the development of synthetic methodology for the direct transformations of amides,16 we reported recently the first general method for the direct reductive alkylation of secondary amides15n and a Tf2O-based17 method for the reduction of amides A to give amines B (Scheme 1).10n The latter report also includes secondary amides 1-1 and 1-2. Herein, we report the results of a systematic investigation on the direct and chemoselective reduction of secondary amides to amines.
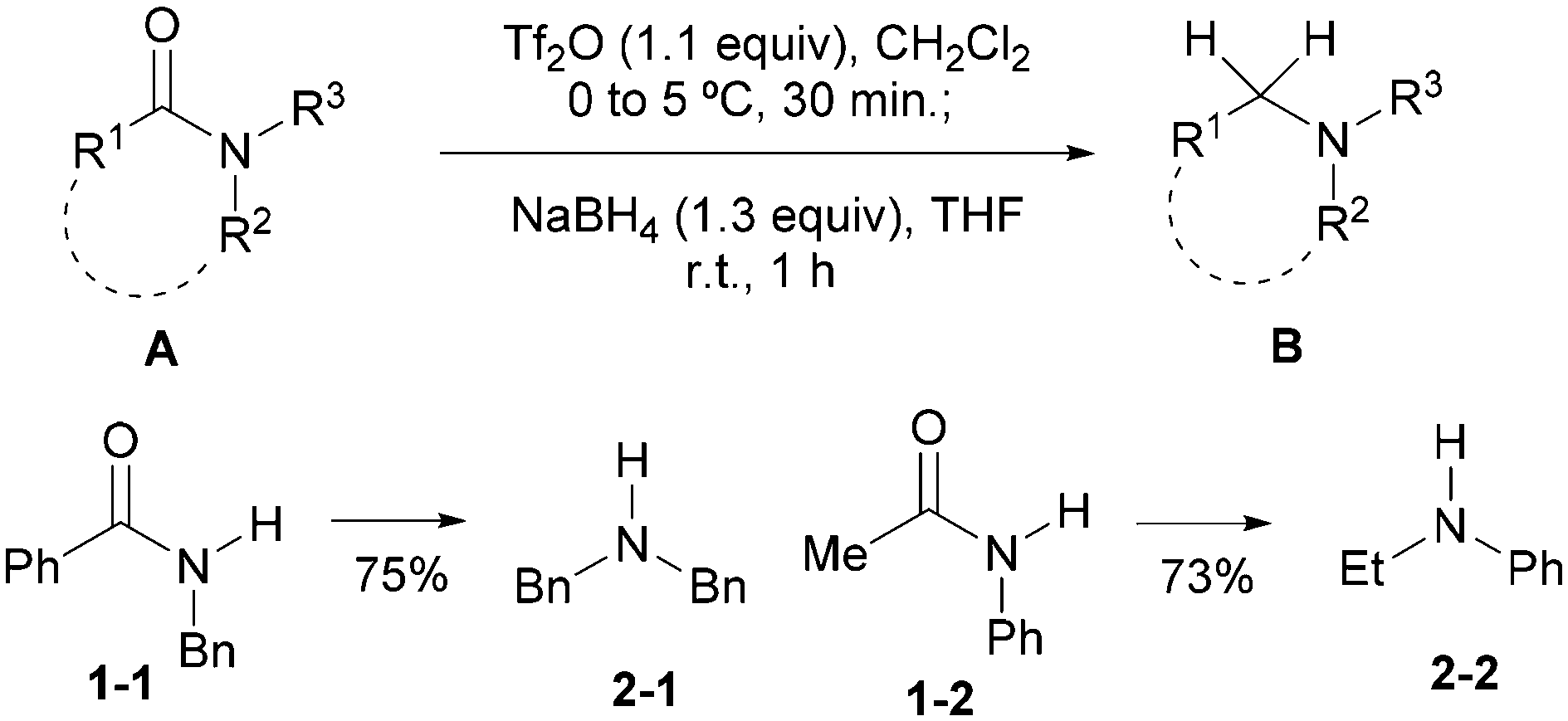 | ||
| Scheme 1 | ||
Results and discussion
In our previous report,10n the optimal conditions defined for the direct reduction of tertiary amides were adapted for the reduction of secondary amides 1-1 and 1-2. Thus in the present investigation, Tf2O and NaBH4 were retained for the in situ amide activation and the reduction of the presumed imidoyl triflate intermediates, while other reaction parameters including a solvent (THF, Et2O, MTBE) and equivalents of sodium borohydride (1.3–2.0 mol equiv.) were re-examined. With Tf2O/CH2Cl2–NaBH4 (1.3 mol equiv.)/THF combination, the reduction of N-i-propylbenzamide 1a gave amine 2a in 74% yield along with 9% of the recovered starting amide (Table 1, entry 1). Next, the effects of 2-fluoropyridine (2-F-Py) were examined. As can be seen from Table 1, use of 0.5 mol equiv. of 2-F-Py (Table 1, entry 2) resulted in an increase of the yield of amine 2a by 5%. However, the yield based on the recovered starting material remained the same. Surprisingly, use of more equiv. of 2-F-Py led to a drop of yield (Table 1, entries 3–5), which is attributed to the competing reduction reaction of the 2-fluoropyridinium ion at a higher concentration. On balancing the yield and atom economy, the optimal conditions for the amide reduction were defined as: Tf2O (1.1 mol equiv.)/CH2Cl2 (0.25 M)–NaBH4 (1.3 mol equiv.)/THF.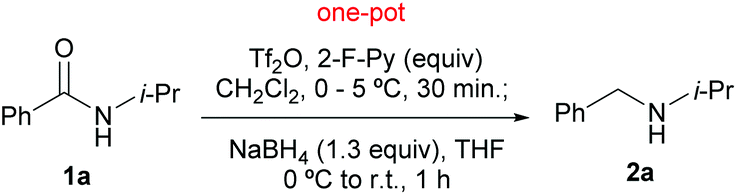
With the optimal conditions in hand, the scope of the reduction was investigated, and the results are summarized in Table 2. We first examined the effects of substituents on the reduction of aromatic amides. As can be seen from entries 1 to 4 and entries 5 to 9 of Table 2, the reactions worked similarly for benzamide derivatives bearing both electron-donating groups (entries 2, 3 and 6) and electron-withdrawing groups (entries 4, 7–9) at the para-position. Slightly lower yields were obtained from substrates bearing an electron donating group. N-i-Propyl and N-c-hexylbenzamides gave similar results (entries 1, 2 versus 5, 6). In addition to ether (entry 3), halo (entries 4, 7 and 8), and CF3 (entry 9) groups, the functional group tolerance of the reaction was further examined. The results shown in entries 10–12 indicate that the reaction also tolerates para-substituted ester, cyano and nitro groups. α,β-Unsaturated amide 1m also reacted chemoselectively to produce the corresponding amine 2m in 65% yield (entry 13). Surprisingly, the reaction of m-bromobenzamide 1n gave the corresponding amine 2n in a lower yield of 64% (entry 14). Good yield was obtained with N-isopropyl-1-naphthamide (entry 15, yield: 78%). It is noteworthy that N-isopropyl-3-phenylpropanamide 1p, an aliphatic amide, also reacted smoothly to give amine 2p in 70% yield (entry 16). However, the reduction of the more hindered N-isopropyl-2-phenylbutanamide 1q afforded the corresponding amine 2q in a modest yield of 63% (entry 17). Interestingly, the reduction of (S)-N-(1-phenylethyl)benzamide 1r produced the corresponding amine 2r in 65% yield (entry 18).
|
|
||
|---|---|---|
| Entry | Substrate (R1, R2) | Product: % yielda |
| a Isolated yield. b Yield based on the recovered starting amide. | ||
| 1 | 1a (Ph, i-Pr) | 2a: 74 (83)b |
| 2 | 1b (p-MeC6H4, i-Pr) | 2b: 67 (74)b |
| 3 | 1c (p-MeOC6H4, i-Pr) | 2c: 71 (79)b |
| 4 | 1d (p-ClC6H4, i-Pr) | 2d: 73 (86)b |
| 5 | 1e (Ph, c-hex) | 2e: 76 (89)b |
| 6 | 1f (p-MeC6H4, c-hex) | 2f: 68 (76)b |
| 7 | 1g (p-BrC6H4, c-hex) | 2g: 75 (87)b |
| 8 | 1h (p-FC6H4, c-hex) | 2h: 73 (82)b |
| 9 | 1i (p-CF3C6H4, c-hex) | 2i: 77 (86)b |
| 10 | 1j (p-MeO2CC6H4, i-Pr) | 2j: 71 (78)b |
| 11 | 1k (p-NCC6H4, i-Pr) | 2k: 72 (91)b |
| 12 | 1l (p-NO2C6H4, i-Pr) | 2l: 76 (89)b |
| 13 | 1m (Phenylethenyl, i-Pr) | 2m: 65 (74)b |
| 14 | 1n (m-BrC6H4, i-Pr) | 2n: 64 (70)b |
| 15 | 1o (1-Naphthyl, i-Pr) | 2o: 78 (86)b |
| 16 | 1p (2-Phenylethyl, i-Pr) | 2p: 70 (70)b |
| 17 | 1q (1-Phenylpropyl, i-Pr) | 2q: 63 (75)b |
| 18 | 1r (Ph, (S)-α-Methylbenzyl) | 2r: 65 (76)b |
| 19 | 1s (Ph, n-Bu) | 2s: 71 (77)b |
| 20 | 1t (Ph, 2-Phenylethyl) | 2t: 72 (81)b |
| 21 | 1u (n-C10H21, Bn) | 2u: 74 (87)b |
| 22 | 1v (2-Thiophenyl, Bn) | 2v: 65 (80)b |
| 23 | 1w (1-Adamantyl, Bn) | 2w: 68 (84)b |
| 24 | 1x (Piperonyl, Bn) | 2x: 56 (62)b |
| 25 | 1y (Ph, Ph) | 2y: Trace (trace)b |
| 26 | 1z (Me, p-MeO2CC6H4) | 2z: Trace (trace)b |
| 27 | 1aa (Ph, t-Bu) | 2aa: Trace (trace)b |
A change of the N-alkyl groups from secondary (i-Pr, c-hex) to primary did not affect the reaction (entries 19 and 20). The reaction of heteroaromatic N-benzylthiophene-2-carboxamide 1v produced amine 2v in 65% yield (entry 22). Even hindered N-benzyladamantane-1-carboxamide 1w reacted smoothly to give amine 2w in 68% yield (entry 23). The reaction also tolerated the acetal group although the corresponding amine was obtained in a modest yield (56%) (piperonamide 1x, entry 24). However, the reaction failed with N-aryl (entries 25 and 26) and N-tert-butyl amides (entry 27). Similarly, attempted selective reduction of picolinamides and 2-furoylamide were unsuccessful.
In view of the widespread use of a secondary amide as a valuable directing group for both lithiation-functionalization and C–H activation-functionalization, we next focused on the application of the present method to the reduction of C–H functionalization products.
The reduction of a Nakamura's C–H functionalization product 514a was first investigated. Under the standard conditions (vide supra), the reduction of 5 proceeded without incident to give 6 in 64% yield (Scheme 2). It is worth mentioning that amide 5 is also a product obtained by C–H lithiation–ethylation.13g,h,14a Compared with the reaction of the parent amide 1a (Table 2, entry 1), the lower yield of amine 6 might be attributed to steric hindrance presented in 5. We next examined the reduction of compound 7, a Daugulis’ C–H functionalization product.14b Subjecting 7 to the conditions that we used previously led to the desired amine 8 in 40% yield. To our delight, the yield improved to 60% by increasing the amount of NaBH4 to 1.5 equiv. and prolonging the reduction time to 12 h. Finally, the method was applied to 9, a Dong's C–H functionalization product.14c As expected, under the standard conditions, the reduction of amide 9 gave amine 10 in 61% yield.
 | ||
| Scheme 2 Reduction of selected C–H functionalization products. | ||
We next examined the reduction of secondary lactams. Attempted reduction of secondary lactam 11, an intermediate in our recent synthesis of alkaloid (−)-streptopyrrolidine,19 to the optimized reduction conditions failed. This is in agreement with our related results.15m The failure led us to recall Ganem's chemistry, who has demonstrated the selective reduction of secondary lactams (2-pyrrolidones) to imines15b,c by Schwartz reagent (Cp2ZrHCl).20 It was envisaged that the presumed imine–zirconium complex could be further reduced by NaBH4. To our delight, treatment of lactam 11 with Cp2ZrHCl in THF at r.t. for 30 min, followed by addition of NaBH4 (3.0 equiv.) smoothly afforded pyrrolidine 12 in an 82% yield. Similar reduction of t-butyl pyroglutamate 13 produced chemoselectively t-butyl prolinate 14 in an 80% yield (Scheme 3). The observed chemoselectivity is in agreement with those observed by Ganem.15d
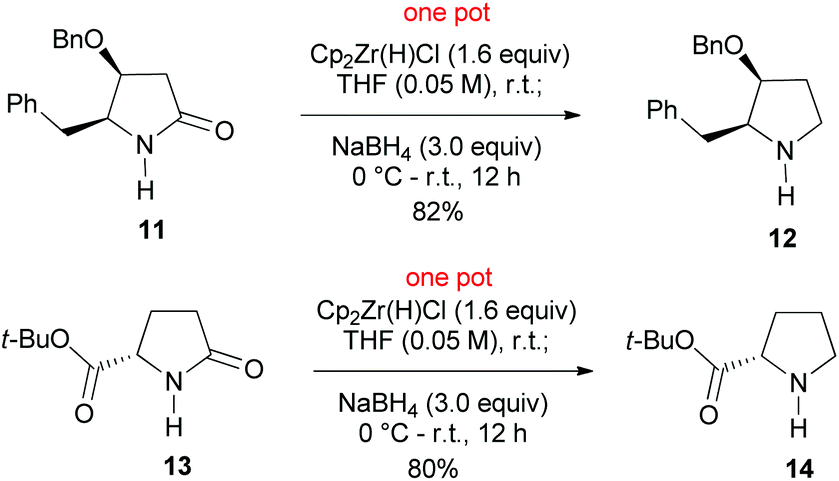 | ||
| Scheme 3 | ||
Conclusion
In summary, on the basis of our preliminary results, we have developed a simple method for the direct reduction of secondary amides, which relied solely on two reagents, Tf2O and NaBH4. Both aromatic and aliphatic amides reacted well, and the N-alkyl group can either be primary or secondary alkyls, but the reaction failed with N-aryl and N-tert-butyl amides. The reaction exhibited good functional group tolerance for a series of reducible functional groups that ranged from halo (Cl, Br, F) to ether (OMe), from ester (CO2Me) to cyano (CN), and from nitro (NO2) to acetal (OCH2O). The reduction of an α,β-unsaturated amide also ran chemoselectively at the carbonyl group. The reduction method is limited to amides. The chemoselective reduction of secondary lactams has been achieved using the Cp2ZrHCl–NaBH4 combination. The functionalized product may be used for further reactions such as coupling reactions. We also demonstrated that the method is applicable to the direct reduction of C–H lithiation–alkylation and C–H functionalization products. To the best of our knowledge, this is the first example of collective and direct reduction of both C–H lithiation-functionalization and C–H functionalization products, which renders the use of directing groups in the chemistry of C–H functionalization more step-economical.Experimental section
General methods
1H NMR and 13C NMR spectra were recorded on a Bruker 400 (1H/400 MHz, 13C/100 MHz) spectrometer. Chemical shifts are expressed in parts per million (δ) relative to an internal standard of residual chloroform (7.26 ppm for 1H NMR and 77.0 ppm for 13C NMR). Data for 1H NMR are reported as chemical shifts (multiplicity, coupling constant, number of protons). ESI-Mass spectra were recorded on a Bruker Dalton ESquire 3000 plus LC-MS apparatus. Optical rotations were measured with an Anton Paar MCP 500 polarimeter. Melting points were determined on a Büchi M560 Automatic Melting Point apparatus. Infrared spectra were recorded with a Nicolet Avatar 330 FT-IR spectrometer using the film or KBr pellet technique.Silica gel (300–400 mesh) was used for flash column chromatography, eluting (unless otherwise stated) with the ethyl-acetate–petroleum ether (PE) (60–90 °C) mixture. Tf2O was distilled over phosphorus pentoxide and used within a week. THF was distilled over sodium benzophenone ketyl under N2. Dichloromethane was distilled over calcium hydride under N2.
General procedure A for the preparation of amines from amides
To a solution of an amide (1.0 mmol) in anhydrous CH2Cl2 (4 mL) was added Tf2O (1.1 mol equiv., 1.1 mmol) in an ice bath. After being stirred for 0.5 h, NaBH4 (1.3 mol equiv., 1.3 mmol) was added in one portion and the mixture was diluted with 2.5 mL of THF. The reaction mixture was stirred for 60 min. The reaction was quenched with H2O (5 mL), and the pH of the reaction mixture was adjusted to 10.5–11.0 by the addition of a saturated aqueous sodium carbonate solution at 0 °C. The cooled aqueous solution was extracted with Et2O (5 × 15 mL). The combined organic layers were washed with brine (5 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel eluting with ethyl acetate/petroleum ether/triethylamine (1%) to afford the amine.General procedure B for the preparation of lactams from amides
A solution of lactam (0.5 mmol) in THF (5 mL) was added dropwise to a suspension of Cp2ZrHCl (1.6 mol equiv., 0.8 mmol, 206 mg) in THF (5 mL) at room temperature under an argon atmosphere. The mixture was stirred until the white suspension disappeared and the solution became clear. Then NaBH4 (3.0 mol equiv., 1.5 mmol) was added in one portion at 0 °C. The mixture was warmed gradually to ambient temperature and stirred overnight. The reaction was quenched with H2O (2 mL), and the pH of the reaction mixture was adjusted to 10.5–11.0 by the addition of a saturated aqueous sodium carbonate solution at 0 °C. The mixture was filtered through a Celite pad (washed with Et2O). The filtrate was extracted with Et2O (5 × 10 mL). The combined organic layers were washed with brine (5 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel eluting with ethyl acetate–petroleum ether to afford the amine.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3), the known amine18a2a (138 mg, yield: 74%) a colourless oil. IR (film) νmax: 3448, 2934, 2856, 1615, 1470, 1367, 1113, 707 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.10 (d, J = 6.2 Hz, 6H, CHCH3), 1.36 (br s, 1H, NH), 2.86 (heptet, J = 6.2 Hz, 1H, NHCH), 3.78 (s, 2H, CH2NH), 7.21–7.27 (m, 1H, Ar–H), 7.28–7.38 (m, 4H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 22.9 (2C), 48.1, 51.6, 126.8, 128.1 (2C), 128.4 (2C), 140.8 ppm; MS (ESI, m/z): 150 (M + H+).
:3), the known amine18a2b (110 mg, yield: 67%) as a pale yellow oil. IR (film) νmax: 3457, 2958, 2930, 2858, 1720, 1664, 1612, 1464, 1272, 1105, 823, 754 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.09 (d, J = 6.2 Hz, 6H, CHCH3), 1.37 (br s, 1H, NH), 2.33 (s, 3H, CH3), 2.84 (heptet, J = 6.2 Hz, 1H, NHCH), 3.74 (s, 2H, CH2NH), 7.12 (d, J = 7.7 Hz, 2H, Ar–H), 7.20 (d, J = 7.7 Hz, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 21.0, 22.9 (2C), 48.0, 51.3, 128.0 (2C), 129.0 (2C), 136.3, 137.7 ppm; MS (ESI, m/z): 164 (M + H+).
:3), the known amine18a,b2c (128 mg, yield: 71%) as a pale yellow oil. IR (film) νmax: 3316, 2967, 2930, 2835, 1615, 1507, 1466, 1242, 1176, 1039, 827 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.09 (d, J = 6.3 Hz, 6H, CHCH3), 1.31 (br s, 1H, NH), 2.84 (heptet, J = 6.3 Hz, 1H, NHCH), 3.74 (s, 2H, CH2NH), 3.79 (s, 3H, OCH3), 6.77–6.94 (m, 2H, Ar–H), 7.19–7.25 (m, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 22.9 (2C), 47.9, 51.0, 55.2, 113.7 (2C), 129.2 (2C), 132.9, 158.5 ppm; MS (ESI, m/z): 180 (M + H+).
:3), the known amine18a2d (134 mg, yield: 73%) as a pale yellow oil. IR (film) νmax: 3316, 2958, 2926, 2847, 1509, 1470, 1379, 1088, 1010, 806 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.09 (d, J = 6.3 Hz, 6H, CHCH3), 1.30 (br s, 1H, NH), 2.83 (heptet, J = 6.3 Hz, 1H, NHCH), 3.74 (s, 2H, CH2NH), 7.19–7.35 (m, 4H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 22.9 (2C), 48.1, 50.8, 128.4 (2C), 129.4 (2C), 132.4, 139.3 ppm; MS (ESI, m/z): 184 (M + H+).
:3), the known amine12b2e (144 mg, yield: 76%) as a colourless oil. IR (film) νmax: 3312, 3025, 2926, 2851, 1603, 1499, 1458, 1122, 1022, 736, 694 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.10–1.31 (m, 5H, c-hex-H), 1.36 (br s, 1H, NH), 1.57–1.65 (m, 1H, c-hex-H), 1.69–1.77 (m, 2H, c-hex -H), 1.90–1.95 (m, 2H, c-hex-H), 2.45–2.52 (m, 1H, NHCH), 3.81 (s, 2H, CH2NH), 7.21–7.25 (m, 1H, Ar–H), 7.31–7.32 (m, 4H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 25.0 (2C), 26.2, 33.6 (2C), 51.0, 56.2, 126.7, 128.1 (2C), 128.4 (2C), 141.0 ppm; MS (ESI, m/z): 190 (M + H+).
:3), the known amine12b2f (138 mg, yield: 68%) as a pale yellow oil. IR (film) νmax: 3316, 2922, 2839, 1520, 1449, 1122, 806, 735 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.09–1.29 (m, 5H, c-hex-H), 1.38 (br s, 1H, NH), 1.58–1.64 (m, 1H, c-hex-H), 1.69–1.76 (m, 2H, c-hex-H), 1.87–1.94 (m, 2H, c-hex-H), 2.33 (s, 3H, CH3), 2.43–2.52 (m, 1H, NHCH), 3.76 (s, 2H, CH2NH), 7.12 (d, J = 7.8 Hz, 2H, Ar–H), 7.20 (d, J = 7.8 Hz, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 21.0, 25.0 (2C), 26.2, 33.5 (2C), 50.7, 56.0, 128.0 (2C), 129.0 (2C), 136.2, 137.9 ppm; MS (ESI, m/z): 204 (M + H+).
:3), the known amine12b2g (101 mg, yield: 75%) as a pale yellow oil. IR (film) νmax: 3303, 2922, 2851, 1478, 1445, 1122, 1072, 806, 737 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.05–1.30 (m, 6H, c-hex-H and NH), 1.60–1.65 (m, 1H, c-hex-H), 1.69–1.77 (m, 2H, c-hex-H), 1.84–1.93 (m, 2H, c-hex-H), 2.41–2.50 (m, 1H, NHCH), 3.76 (s, 2H, CH2NH), 7.17–7.24 (m, 2H, Ar–H), 7.41–7.45 (m, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 25.0 (2C), 26.1, 33.6 (2C), 50.3, 56.1, 120.4, 129.7 (2C), 131.4 (2C), 140.1 ppm; MS (ESI, m/z): 268 and 270 (M + H+).
:3), the known amine12b2h (76 mg, yield: 73%) as a pale yellow oil. IR (film) νmax: 3299, 2934, 2851, 1611, 1507, 1449, 1221, 1118, 835 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.03–1.33 (m, 6H, c-hex-H and NH), 1.59–1.65 (m, 1H, c-hex-H), 1.70–1.77 (m, 2H, c-hex-H), 1.87–1.95 (m, 2H, c-hex-H), 2.41–2.50 (m, 1H, NHCH), 3.77 (s, 2H, CH2NH), 6.90–7.04 (m, 2H, Ar–H), 7.24–7.32 (m, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 25.0 (2C), 26.1, 33.6 (2C), 50.3, 56.2, 115.1 (2C) (d, 2J(C,F) = 21.3 Hz), 129.5 (2C) (d, 3J(C,F) = 8.0 Hz), 136.7, 161.8 (d, 1J(C,F) = 244.4 Hz) ppm; MS (ESI, m/z): 208 (M + H+).
:3), the known amine12b2i (97 mg, yield: 77%) as a yellow oil. IR (film) νma: 3307, 2934, 2868, 1615, 1453, 1325, 1158, 1122, 845, 823 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.03–1.30 (m, 6H, c-hex-H and NH), 1.59–1.67 (m, 1H, c-hex-H), 1.69–1.79 (m, 2H, c-hex-H), 1.87–1.96 (m, 2H, c-hex-H), 2.43–2.52 (m, 1H, NHCH), 3.87 (s, 2H, CH2NH), 7.44 (d, J = 8.0 Hz, 2H, Ar–H), 7.57 (d, J = 8.0 Hz, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 24.9 (2C), 26.1, 33.6 (2C), 50.5, 56.2, 124.3 (d, 1J(C,F) = 271.7 Hz), 125.2 (q, 3J(C,F) = 3.7 Hz) (2C), 128.2 (2C), 129.0 (d, 2J(C,F) = 32.2 Hz), 145.2 ppm; MS (ESI, m/z): 258 (M + H+).
:3), amine 2j (147 mg, yield: 71%) as a pale yellow oil. IR (film) νmax: 3312, 2971, 2880, 1731, 1603, 1433, 1267, 1105, 1026, 761 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.10 (d, J = 6.3 Hz, 6H, CHCH3), 1.41 (br s, 1H, NH), 2.84 (heptet, J = 6.3 Hz, 1H, NHCH), 3.84 (s, 2H, CH2NH), 3.90 (s, 3H, COOCH3), 7.40 (d, J = 8.0 Hz, 2H, Ar–H), 7.99 (d, J = 8.2 Hz, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 22.8 (2C), 48.2, 51.2, 51.9, 127.9 (2C), 128.6, 129.6 (2C), 146.2, 167.0 ppm; HRMS (ESI, m/z) calcd for [C12H18NO]+ (M + H+): 208.1338; found: 208.1336.
:3), amine 2k (126 mg, yield: 72%) as a pale yellow oil. IR (film) νmax: 3316, 2959, 2929, 2868, 2225, 1608, 1467, 1380, 1174, 1125, 1022, 825 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.10 (d, J = 6.2 Hz, 6H, CHCH3), 1.36 (br s, 1H, NH), 2.84 (heptet, J = 6.3 Hz, 1H, NHCH), 3.85 (s, 2H, CH2NH), 7.45 (d, J = 8.1 Hz, 2H, Ar–H), 7.61 (d, J = 8.1 Hz, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 22.9 (2C), 48.3, 51.0, 110.6, 119.0, 128.6 (2C), 132.1 (2C), 146.6 ppm; HRMS (ESI, m/z) calcd for [C11H15N2]+ (M + H+): 175.1235; found: 175.1232.
:3), amine 2l (148 mg, yield: 76%) as a yellow oil. IR (film) νmax: 3316, 2967, 2855, 1611, 1520, 1350, 1101, 802, 736 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.11 (d, J = 6.3 Hz, 6H, CHCH3), 1.35 (br s, 1H, NH), 2.85 (heptet, J = 6.3 Hz, 1H, NHCH), 3.90 (s, 2H, CH2NH), 7.51 (d, J = 8.7 Hz, 2H, Ar–H), 8.17 (d, J = 8.7 Hz, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 22.9, 48.4, 50.8, 123.5, 128.6, 146.9, 148.8 ppm; HRMS (ESI, m/z) calcd for [C10H15N2O2]+ (M + H+): 195.1134; found: 195.1135.
:3), the known amine18d2m (114 mg, yield: 65%) as a pale yellow oil. IR (film) νmax: 3370, 2967, 2922, 2855, 1565, 1458, 1383, 977, 748, 690 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.12 (d, J = 6.3 Hz, 6H, CHCH3), 2.93 (heptet, J = 6.3 Hz, 1H, NHCH), 3.25 (br s, 1H, NH), 3.44 (dd, J = 6.5, 1.2 Hz, 2H, CH2NH), 6.31 (dt, J = 15.8, 6.5 Hz, 1H, CH
:3), the known amine18a2a (138 mg, yield: 74%) a colourless oil. IR (film) νmax: 3448, 2934, 2856, 1615, 1470, 1367, 1113, 707 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.10 (d, J = 6.2 Hz, 6H, CHCH3), 1.36 (br s, 1H, NH), 2.86 (heptet, J = 6.2 Hz, 1H, NHCH), 3.78 (s, 2H, CH2NH), 7.21–7.27 (m, 1H, Ar–H), 7.28–7.38 (m, 4H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 22.9 (2C), 48.1, 51.6, 126.8, 128.1 (2C), 128.4 (2C), 140.8 ppm; MS (ESI, m/z): 150 (M + H+).
:3), the known amine18a2b (110 mg, yield: 67%) as a pale yellow oil. IR (film) νmax: 3457, 2958, 2930, 2858, 1720, 1664, 1612, 1464, 1272, 1105, 823, 754 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.09 (d, J = 6.2 Hz, 6H, CHCH3), 1.37 (br s, 1H, NH), 2.33 (s, 3H, CH3), 2.84 (heptet, J = 6.2 Hz, 1H, NHCH), 3.74 (s, 2H, CH2NH), 7.12 (d, J = 7.7 Hz, 2H, Ar–H), 7.20 (d, J = 7.7 Hz, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 21.0, 22.9 (2C), 48.0, 51.3, 128.0 (2C), 129.0 (2C), 136.3, 137.7 ppm; MS (ESI, m/z): 164 (M + H+).
:3), the known amine18a,b2c (128 mg, yield: 71%) as a pale yellow oil. IR (film) νmax: 3316, 2967, 2930, 2835, 1615, 1507, 1466, 1242, 1176, 1039, 827 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.09 (d, J = 6.3 Hz, 6H, CHCH3), 1.31 (br s, 1H, NH), 2.84 (heptet, J = 6.3 Hz, 1H, NHCH), 3.74 (s, 2H, CH2NH), 3.79 (s, 3H, OCH3), 6.77–6.94 (m, 2H, Ar–H), 7.19–7.25 (m, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 22.9 (2C), 47.9, 51.0, 55.2, 113.7 (2C), 129.2 (2C), 132.9, 158.5 ppm; MS (ESI, m/z): 180 (M + H+).
:3), the known amine18a2d (134 mg, yield: 73%) as a pale yellow oil. IR (film) νmax: 3316, 2958, 2926, 2847, 1509, 1470, 1379, 1088, 1010, 806 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.09 (d, J = 6.3 Hz, 6H, CHCH3), 1.30 (br s, 1H, NH), 2.83 (heptet, J = 6.3 Hz, 1H, NHCH), 3.74 (s, 2H, CH2NH), 7.19–7.35 (m, 4H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 22.9 (2C), 48.1, 50.8, 128.4 (2C), 129.4 (2C), 132.4, 139.3 ppm; MS (ESI, m/z): 184 (M + H+).
:3), the known amine12b2e (144 mg, yield: 76%) as a colourless oil. IR (film) νmax: 3312, 3025, 2926, 2851, 1603, 1499, 1458, 1122, 1022, 736, 694 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.10–1.31 (m, 5H, c-hex-H), 1.36 (br s, 1H, NH), 1.57–1.65 (m, 1H, c-hex-H), 1.69–1.77 (m, 2H, c-hex -H), 1.90–1.95 (m, 2H, c-hex-H), 2.45–2.52 (m, 1H, NHCH), 3.81 (s, 2H, CH2NH), 7.21–7.25 (m, 1H, Ar–H), 7.31–7.32 (m, 4H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 25.0 (2C), 26.2, 33.6 (2C), 51.0, 56.2, 126.7, 128.1 (2C), 128.4 (2C), 141.0 ppm; MS (ESI, m/z): 190 (M + H+).
:3), the known amine12b2f (138 mg, yield: 68%) as a pale yellow oil. IR (film) νmax: 3316, 2922, 2839, 1520, 1449, 1122, 806, 735 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.09–1.29 (m, 5H, c-hex-H), 1.38 (br s, 1H, NH), 1.58–1.64 (m, 1H, c-hex-H), 1.69–1.76 (m, 2H, c-hex-H), 1.87–1.94 (m, 2H, c-hex-H), 2.33 (s, 3H, CH3), 2.43–2.52 (m, 1H, NHCH), 3.76 (s, 2H, CH2NH), 7.12 (d, J = 7.8 Hz, 2H, Ar–H), 7.20 (d, J = 7.8 Hz, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 21.0, 25.0 (2C), 26.2, 33.5 (2C), 50.7, 56.0, 128.0 (2C), 129.0 (2C), 136.2, 137.9 ppm; MS (ESI, m/z): 204 (M + H+).
:3), the known amine12b2g (101 mg, yield: 75%) as a pale yellow oil. IR (film) νmax: 3303, 2922, 2851, 1478, 1445, 1122, 1072, 806, 737 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.05–1.30 (m, 6H, c-hex-H and NH), 1.60–1.65 (m, 1H, c-hex-H), 1.69–1.77 (m, 2H, c-hex-H), 1.84–1.93 (m, 2H, c-hex-H), 2.41–2.50 (m, 1H, NHCH), 3.76 (s, 2H, CH2NH), 7.17–7.24 (m, 2H, Ar–H), 7.41–7.45 (m, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 25.0 (2C), 26.1, 33.6 (2C), 50.3, 56.1, 120.4, 129.7 (2C), 131.4 (2C), 140.1 ppm; MS (ESI, m/z): 268 and 270 (M + H+).
:3), the known amine12b2h (76 mg, yield: 73%) as a pale yellow oil. IR (film) νmax: 3299, 2934, 2851, 1611, 1507, 1449, 1221, 1118, 835 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.03–1.33 (m, 6H, c-hex-H and NH), 1.59–1.65 (m, 1H, c-hex-H), 1.70–1.77 (m, 2H, c-hex-H), 1.87–1.95 (m, 2H, c-hex-H), 2.41–2.50 (m, 1H, NHCH), 3.77 (s, 2H, CH2NH), 6.90–7.04 (m, 2H, Ar–H), 7.24–7.32 (m, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 25.0 (2C), 26.1, 33.6 (2C), 50.3, 56.2, 115.1 (2C) (d, 2J(C,F) = 21.3 Hz), 129.5 (2C) (d, 3J(C,F) = 8.0 Hz), 136.7, 161.8 (d, 1J(C,F) = 244.4 Hz) ppm; MS (ESI, m/z): 208 (M + H+).
:3), the known amine12b2i (97 mg, yield: 77%) as a yellow oil. IR (film) νma: 3307, 2934, 2868, 1615, 1453, 1325, 1158, 1122, 845, 823 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.03–1.30 (m, 6H, c-hex-H and NH), 1.59–1.67 (m, 1H, c-hex-H), 1.69–1.79 (m, 2H, c-hex-H), 1.87–1.96 (m, 2H, c-hex-H), 2.43–2.52 (m, 1H, NHCH), 3.87 (s, 2H, CH2NH), 7.44 (d, J = 8.0 Hz, 2H, Ar–H), 7.57 (d, J = 8.0 Hz, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 24.9 (2C), 26.1, 33.6 (2C), 50.5, 56.2, 124.3 (d, 1J(C,F) = 271.7 Hz), 125.2 (q, 3J(C,F) = 3.7 Hz) (2C), 128.2 (2C), 129.0 (d, 2J(C,F) = 32.2 Hz), 145.2 ppm; MS (ESI, m/z): 258 (M + H+).
:3), amine 2j (147 mg, yield: 71%) as a pale yellow oil. IR (film) νmax: 3312, 2971, 2880, 1731, 1603, 1433, 1267, 1105, 1026, 761 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.10 (d, J = 6.3 Hz, 6H, CHCH3), 1.41 (br s, 1H, NH), 2.84 (heptet, J = 6.3 Hz, 1H, NHCH), 3.84 (s, 2H, CH2NH), 3.90 (s, 3H, COOCH3), 7.40 (d, J = 8.0 Hz, 2H, Ar–H), 7.99 (d, J = 8.2 Hz, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 22.8 (2C), 48.2, 51.2, 51.9, 127.9 (2C), 128.6, 129.6 (2C), 146.2, 167.0 ppm; HRMS (ESI, m/z) calcd for [C12H18NO]+ (M + H+): 208.1338; found: 208.1336.
:3), amine 2k (126 mg, yield: 72%) as a pale yellow oil. IR (film) νmax: 3316, 2959, 2929, 2868, 2225, 1608, 1467, 1380, 1174, 1125, 1022, 825 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.10 (d, J = 6.2 Hz, 6H, CHCH3), 1.36 (br s, 1H, NH), 2.84 (heptet, J = 6.3 Hz, 1H, NHCH), 3.85 (s, 2H, CH2NH), 7.45 (d, J = 8.1 Hz, 2H, Ar–H), 7.61 (d, J = 8.1 Hz, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 22.9 (2C), 48.3, 51.0, 110.6, 119.0, 128.6 (2C), 132.1 (2C), 146.6 ppm; HRMS (ESI, m/z) calcd for [C11H15N2]+ (M + H+): 175.1235; found: 175.1232.
:3), amine 2l (148 mg, yield: 76%) as a yellow oil. IR (film) νmax: 3316, 2967, 2855, 1611, 1520, 1350, 1101, 802, 736 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.11 (d, J = 6.3 Hz, 6H, CHCH3), 1.35 (br s, 1H, NH), 2.85 (heptet, J = 6.3 Hz, 1H, NHCH), 3.90 (s, 2H, CH2NH), 7.51 (d, J = 8.7 Hz, 2H, Ar–H), 8.17 (d, J = 8.7 Hz, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 22.9, 48.4, 50.8, 123.5, 128.6, 146.9, 148.8 ppm; HRMS (ESI, m/z) calcd for [C10H15N2O2]+ (M + H+): 195.1134; found: 195.1135.
:3), the known amine18d2m (114 mg, yield: 65%) as a pale yellow oil. IR (film) νmax: 3370, 2967, 2922, 2855, 1565, 1458, 1383, 977, 748, 690 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.12 (d, J = 6.3 Hz, 6H, CHCH3), 2.93 (heptet, J = 6.3 Hz, 1H, NHCH), 3.25 (br s, 1H, NH), 3.44 (dd, J = 6.5, 1.2 Hz, 2H, CH2NH), 6.31 (dt, J = 15.8, 6.5 Hz, 1H, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2), 6.54 (dt, J = 15.8, 1.2 Hz, 1H, PhCHCHCH2), 7.18–7.26 (m, 1H, Ar–H), 7.27–7.34 (m, 2H, Ar–H), 7.34–7.42 (m, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 22.5 (2C), 48.1, 49.1, 126.3 (2C), 127.4, 127.8, 128.5 (2C), 131.7, 137.0 ppm; MS (ESI, m/z): 176 (M + H+).
:3), amine 2n (73 mg, yield: 64%) as a pale yellow oil. IR (film) νmax: 3324, 3071, 2959, 2927, 2865, 1599, 1569, 1126, 1067, 776, 690 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.10 (d, J = 6.3 Hz, 6H, CHCH3), 1.42 (br s, 1H, NH), 2.84 (heptet, J = 6.3 Hz, 1H, NHCH), 3.75 (s, 2H, CH2NH), 7.15–7.21 (m, 1H, Ar–H), 7.22–7.26 (m, 1H, Ar–H), 7.34–7.40 (m, 1H, Ar–H), 7.46–7.52 (m, 1H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 22.9 (2C), 48.1, 51.0, 122.5, 126.6, 129.9 (2C), 131.1, 143.3 ppm; HRMS (ESI, m/z) calcd for [C10H15BrN]+ (M + H+): 228.0388 and 230.0367; found: 228.0389 and 230.0369.
:3), the known amine18b2o (156 mg, yield: 78%) as a pale yellow oil. IR (film) νmax: 3316, 3045, 2963, 2928, 2868, 2825, 1510, 1467, 1126, 1396, 1171, 802, 792, 777 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.15 (d, J = 6.3 Hz, 6H, CHCH3), 1.39 (br s, 1H, NH), 2.97 (heptet, J = 6.3 Hz, 1H, NHCH), 4.21 (s, 2H, CH2NH), 7.36–7.44 (m, 1H, Ar–H), 7.44–7.56 (m, 3H, Ar–H), 7.73–7.77 (m, 1H, Ar–H), 7.82–7.87 (m, 1H, Ar–H), 8.08–8.15 (m, 1H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 23.0 (2C), 48.9, 49.3, 123.5, 125.4, 125.5, 126.0 (2C), 127.6, 128.7, 131.7, 133.8, 136.3 ppm; MS (ESI, m/z): 200 (M + H+).
:3), the known amine11e2p (124 mg, yield: 70%) as a colourless oil. IR (film) νmax: 3312, 2967, 2835, 1578, 1569, 1112, 1014, 803, 694 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.36 (d, J = 6.5 Hz, 6H, CHCH3), 2.16–2.25 (m, 2H, CH2CH2CH2), 2.65 (t, J = 7.5 Hz, 2H, CH2CH2CH2), 2.81–2.87 (m, 2H, CH2CH2NH), 3.20 (heptet, J = 6.5 Hz, 1H, NHCH), 7.15–7.20 (m, 3H, Ar–H), 7.24–7.29 (m, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 19.4 (2C), 27.8, 32.9, 44.3, 50.2, 126.2, 128.3 (2C), 128.5 (2C), 140.1 ppm; MS (ESI, m/z): 178 (M + H+).
:2), amine 2q (121 mg, yield: 63%) as a pale yellow oil. IR (film) νmax: 3436, 2958, 2917, 2849, 1578, 1462, 1258, 1114, 1012, 802, 694 cm−1; 1H NMR (CDCl3, 400 MHz): δ 0.80 (t, J = 7.5 Hz, 3H), 0.95 (d, J = 6.3 Hz, 3H), 1.00 (d, J = 6.3 Hz, 3H, CH3CH2), 1.19 (br s, 1H, NH), 1.51–1.60 (m, 1H, CH3CH2), 1.66–1.75 (m, 1H, CH3CH2), 2.62–2.78 (m, 3H, CH2NH and CH2CHCH2), 2.85–2.93 (m, 1H, NHCH), 7.14–7.25 (m, 3H, Ar–H), 7.27–7.33 (m, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 12.0, 22.7, 22.8, 27.6, 47.9, 48.5, 53.0, 126.3, 127.8 (2C), 128.4 (2C), 143.8 ppm; HRMS (ESI, m/z) calcd for [C13H22N]+ (M + H+): 192.1752; found: 192.1750.
:3), the known amine18e2r (137 mg, yield: 65%) as a pale yellow oil. IR (film) νmax: 3332, 3029, 2959, 2926, 2843, 1603, 1503, 1449, 1358, 761, 699 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.37 (dd, J = 6.6, 0.8 Hz, 3H, CHCH3), 1.71 (br s, 1H, NH), 3.59 (d, J = 13.1 Hz, 1H, CH2NH), 3.66 (d, J = 13.1 Hz, 1H, CH2NH), 3.81 (q, J = 6.7 Hz, 1H, NHCH), 7.20–7.40 (m, 10H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 24.5, 51.6, 57.5, 126.7 (2C), 126.8, 126.9, 128.1 (2C), 128.3 (2C), 128.4 (2C), 140.6, 145.5 ppm; MS (ESI, m/z): 212 (M + H+).
:3), the known amine18f2s (116 mg, yield: 71%) as a pale yellow oil. IR (film) νmax: 3316, 3063, 2955, 2918, 2851, 1603, 1499, 1462, 1130, 740, 694 cm−1; 1H NMR (CDCl3, 400 MHz): δ 0.91 (t, J = 7.3 Hz, 3H, CH2CH3), 1.30–1.40 (m, 2H, CH2CH2CH3), 1.43 (br s, 1H, NH), 1.46–1.54 (m, 2H, CH2CH2CH3), 2.63 (t, J = 7.3 Hz, 2H, NHCH2CH2), 3.78 (s, 2H, CH2NH), 7.21–7.28 (m, 1H, Ar–H), 7.28–7.34 (m, 4H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 14.0, 20.4, 32.2, 49.2, 54.1, 126.8, 128.1 (2C), 128.3 (2C), 140.5 ppm; MS (ESI, m/z): 164 (M + H+).
:3), the known amine12d2t (153 mg, yield: 72%) as a pale yellow oil. IR (film) νmax: 3324, 3063, 3021, 2926, 2851, 2818, 1599, 1503, 1449, 1122, 1035, 753, 694 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.55 (br s, 1H, NH), 2.80–2.86 (m, 2H, NHCH2CH2), 2.88–2.93 (m, 2H, NHCH2CH2), 3.80 (s, 2H, CH2NH), 7.17–7.25 (m, 4H, Ar–H), 7.25–7.33 (m, 6H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 36.3, 50.5, 53.8, 126.1, 126.9, 128.0 (2C), 128.3 (2C), 128.4 (2C), 128.7 (2C), 140.0, 140.2 ppm; MS (ESI, m/z): 212 (M + H+).
:3), the known amine18d2u (192 mg, yield: 74%) as a colourless oil. IR (film) νmax: 3320, 2934, 2851, 1487, 1458, 1122, 732, 693 cm−1; 1H NMR (CDCl3, 400 MHz): δ 0.88 (t, J = 7.0 Hz, 3H, CH2CH3), 1.23–1.32 (m, 16H, CH2(CH2)8CH2), 1.42 (br s, 1H, NH), 1.47–1.54 (m, 2H, CH2CH2CH3), 2.62 (t, J = 7.3 Hz, 2H, NHCH2CH2), 3.78 (s, 2H, NHCH2), 7.22–7.27 (m, 1H, Ar–H), 7.32 (m, 4H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 14.1, 22.7, 27.4, 29.3, 29.6 (4C), 30.1, 31.9, 49.5, 54.1, 126.8, 128.1 (2C), 128.3 (2C), 140.6 ppm; MS (ESI, m/z): 262 (M + H+).
:3), the known amine12e2v (66 mg, yield: 65%) as a pale yellow oil. IR (film) νmax: 3320, 3026, 2919, 2849, 1453, 1263, 1107, 735, 699 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.76 (br s, 1H, NH), 3.83 (s, 2H, NHCH2), 3.99 (s, 2H, CH2NH), 6.87–7.00 (m, 2H, Ar–H), 7.20–7.28 (m, 2H, Ar–H), 7.29–7.39 (m, 4H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 47.5, 52.7, 124.3, 124.8, 126.6, 127.0, 128.2 (2C), 128.4 (2C), 140.0, 144.2 ppm; MS (ESI, m/z): 204 (M + H+).
:3), amine 2w (87 mg, yield: 68%) as a pale yellow oil. IR (film) νmax: 3361, 3026, 2905, 2846, 1494, 1452, 1107, 733, 697 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.51–1.55 (m, 6H, CHCH2CH and NH), 1.61–1.67 (m, 4H, CHCH2CH), 1.69–1.74 (m, 3H, CHCH2CH), 1.97 (m, 3H, CH2CHCH2), 2.25 (s, 2H, CH2NH), 3.79 (s, 2H, NHCH2), 7.21–7.26 (m, 1H, Ar–H), 7.29–7.35 (m, 4H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 28.5 (3C), 33.5, 37.3 (3C), 40.9 (3C), 54.7, 62.2, 126.7, 127.9 (2C), 128.3 (2C), 141.0 ppm; HRMS (ESI, m/z) calcd for [C18H26N]+ (M + H+): 256.2065; found: 256.2061.
:3), the known amine12e2x (68 mg, yield: 56%) as a pale yellow oil. IR (film) νmax: 3336, 3021, 2916, 2849, 1607, 1501, 1495, 1441, 1246, 807, 736, 698 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.64 (br s, 1H, NH), 3.71 (s, 2H, CH2NH), 3.78 (s, 2H, NHCH2), 5.92 (s, 2H, OCH2O), 6.73–6.80 (m, 2H, Ar–H), 6.86 (s, 1H, Ar–H), 7.23–7.28 (m, 1H, Ar–H), 7.29–7.37 (m, 4H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 52.9 (2C), 100.8, 108.0, 108.7, 121.2, 126.9, 128.1 (2C), 128.4 (2C), 134.3, 140.3, 146.5, 147.7 ppm; MS (ESI, m/z): 242 (M + H+).
:3), amine 6 (56 mg, yield: 64%) as a pale yellow oil. IR (film) νmax: 3312, 2971, 2934, 2855, 1462, 1453, 1375, 1179, 735 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.12 (d, J = 6.3 Hz, 6H, CHCH3), 1.24 (t, J = 7.6 Hz, 3H, CH2CH3), 1.36 (br s, 1H, NH), 2.71 (q, J = 7.6 Hz, 2H, CH2CH3), 2.89 (heptet, J = 6.3 Hz, 1H, NHCH), 3.77 (s, 2H, CH2NH), 7.14–7.22 (m, 3H, Ar–H), 7.30 (d, J = 7.1 Hz, 1H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 15.4, 23.0 (2C), 25.3, 48.7, 48.8, 125.9, 127.1, 128.5, 128.8, 138.0, 142.2 ppm; HRMS (ESI, m/z) calcd for [C12H20N]+ (M + H+): 178.1596; found: 178.1596.
:3), amine 8 (116 mg, yield: 60%) as a pale yellow oil. IR (film) νmax: 3324, 3054, 2965, 2929, 1610, 1588, 1470, 1375, 1259, 1168, 794, 702, 636 cm−1; 1H NMR (CDCl3, 400 MHz): δ 0.96 (d, J = 6.3 Hz, 6H, CHCH3), 1.36 (br s, 1H, NH), 2.67 (heptet, J = 6.3 Hz, 1H, NHCH), 3.64 (s, 2H, CH2NH), 7.10 (d, J = 8.1 Hz, 1H, Ar–H), 7.22–7.26 (m, 1H, Ar–H), 7.28–7.33 (m, 2H, Ar–H), 7.41–7.48 (m, 2H, Ar–H), 7.64 (m, 1H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 22.7 (2C), 48.4, 48.7, 119.2, 119.8, 121.6, 121.8, 122.2, 127.4, 129.6, 130.1, 131.4, 132.3, 139.0, 140.3, 142.1, 149.1 ppm; HRMS (ESI, m/z) calcd for [C17H18BrF3NO]+ (M + H+): 388.0524 and 390.0503; found: 388.0512 and 390.0503.
:3), amine 10 (96 mg, yield: 61%) as a pale yellow oil. IR (film) νmax: 3328, 3056, 2962, 2867, 1499, 1457, 1171, 760, 702 cm−1; 1H NMR (CDCl3, 400 MHz): δ 0.69 (d, J = 6.3 Hz, 6H, CHCH3), 0.91 (br s, 1H, NH), 2.28–2.40 (m, 3H, CH2CH2 and NH), 2.69–2.76 (m, 2H, CH2CH2NH), 7.16–7.22 (m, 2H, Ar–H), 7.23–7.29 (m, 1H, Ar–H), 7.31–7.44 (m, 10H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 22.6 (2C), 30.9, 47.1, 47.2, 125.3, 126.9 (2C), 128.1 (4C), 129.2 (4C), 129.4 (2C), 135.3 (2C), 142.2 (2C), 142.9 ppm; HRMS (ESI, m/z) calcd for [C23H26N]+ (M + H+): 316.2065; found: 316.2062.
:5), pyrrolidine 12 (80 mg, yield: 82%) as a colourless oil. [α]20D +152.7 (c 1.0, CHCl3); IR (film) νmax: 3440, 3034, 2926, 2872, 1495, 1453, 1180, 1047, 740, 707 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.84–1.93 (m, 1H, OCHCH2CH2), 2.00–2.09 (m, 1H, OCHCH2CH2), 2.87–3.14 (m, 3H, NHCH2 and PhCH2CH), 3.38–3.46 (dd, J = 12.1, 3.1 Hz, 1H, PhCH2CH), 3.46–3.56 (m, 1H, NHCH), 3.79 (t, J = 3.4 Hz, 1H, OCHCH2), 3.98 (br s, 1H, NH), 4.30 (d, J = 11.3 Hz, 1H, OCH2Ph), 4.50 (d, J = 11.3 Hz, 1H, OCH2Ph), 7.16–7.43 (m, 10H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 27.7, 31.9, 53.0, 70.6, 72.3, 77.7, 126.6, 127.8, 128.0, 128.6 (2C), 129.1, 137.4, 137.9 ppm; HRMS (ESI, m/z) calcd for [C18H21NNaO]+ (M + Na+): 290.1521; found: 290.1510.
:20), the known proline derivative18g,h14 (75 mg, yield: 80%) as a white solid. Mp: 88–89 °C (EtOAc–hexane); [α]20D −47.6 (c 1.0, CHCl3 {lit.18i [α]24D −40.5 (c 0.4, MeOH)}. IR (film) νmax: 3443, 2980, 2930, 1727, 1379, 1180, 1159 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.49 (s, 9H, CH3), 1.75–1.83 (m, 1H, CH2CH2CH2), 1.89–1.98 (m, 1H, CH2CH2CH2), 2.00–2.11 (m, 1H, CH2CH2CH), 2.29–2.41 (m, 1H, CH2CH2CH), 2.87–2.98 (m, 1H, CH2CH2NH), 3.33–3.42 (m, 1H, CH2CH2NH), 3.63–3.71 (m, 1H, COCHNH), 4.98 (br s, 1H, NH) ppm; 13C NMR (CDCl3, 100 MHz): δ 24.8, 27.9 (3C), 30.0, 54.9, 67.3, 83.6, 171.2 ppm; HRMS (ESI, m/z) calcd for [C9H17NNaO2]+ (M + Na+): 194.1157; found: 194.1150.
CHCH2), 6.54 (dt, J = 15.8, 1.2 Hz, 1H, PhCHCHCH2), 7.18–7.26 (m, 1H, Ar–H), 7.27–7.34 (m, 2H, Ar–H), 7.34–7.42 (m, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 22.5 (2C), 48.1, 49.1, 126.3 (2C), 127.4, 127.8, 128.5 (2C), 131.7, 137.0 ppm; MS (ESI, m/z): 176 (M + H+).
:3), amine 2n (73 mg, yield: 64%) as a pale yellow oil. IR (film) νmax: 3324, 3071, 2959, 2927, 2865, 1599, 1569, 1126, 1067, 776, 690 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.10 (d, J = 6.3 Hz, 6H, CHCH3), 1.42 (br s, 1H, NH), 2.84 (heptet, J = 6.3 Hz, 1H, NHCH), 3.75 (s, 2H, CH2NH), 7.15–7.21 (m, 1H, Ar–H), 7.22–7.26 (m, 1H, Ar–H), 7.34–7.40 (m, 1H, Ar–H), 7.46–7.52 (m, 1H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 22.9 (2C), 48.1, 51.0, 122.5, 126.6, 129.9 (2C), 131.1, 143.3 ppm; HRMS (ESI, m/z) calcd for [C10H15BrN]+ (M + H+): 228.0388 and 230.0367; found: 228.0389 and 230.0369.
:3), the known amine18b2o (156 mg, yield: 78%) as a pale yellow oil. IR (film) νmax: 3316, 3045, 2963, 2928, 2868, 2825, 1510, 1467, 1126, 1396, 1171, 802, 792, 777 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.15 (d, J = 6.3 Hz, 6H, CHCH3), 1.39 (br s, 1H, NH), 2.97 (heptet, J = 6.3 Hz, 1H, NHCH), 4.21 (s, 2H, CH2NH), 7.36–7.44 (m, 1H, Ar–H), 7.44–7.56 (m, 3H, Ar–H), 7.73–7.77 (m, 1H, Ar–H), 7.82–7.87 (m, 1H, Ar–H), 8.08–8.15 (m, 1H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 23.0 (2C), 48.9, 49.3, 123.5, 125.4, 125.5, 126.0 (2C), 127.6, 128.7, 131.7, 133.8, 136.3 ppm; MS (ESI, m/z): 200 (M + H+).
:3), the known amine11e2p (124 mg, yield: 70%) as a colourless oil. IR (film) νmax: 3312, 2967, 2835, 1578, 1569, 1112, 1014, 803, 694 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.36 (d, J = 6.5 Hz, 6H, CHCH3), 2.16–2.25 (m, 2H, CH2CH2CH2), 2.65 (t, J = 7.5 Hz, 2H, CH2CH2CH2), 2.81–2.87 (m, 2H, CH2CH2NH), 3.20 (heptet, J = 6.5 Hz, 1H, NHCH), 7.15–7.20 (m, 3H, Ar–H), 7.24–7.29 (m, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 19.4 (2C), 27.8, 32.9, 44.3, 50.2, 126.2, 128.3 (2C), 128.5 (2C), 140.1 ppm; MS (ESI, m/z): 178 (M + H+).
:2), amine 2q (121 mg, yield: 63%) as a pale yellow oil. IR (film) νmax: 3436, 2958, 2917, 2849, 1578, 1462, 1258, 1114, 1012, 802, 694 cm−1; 1H NMR (CDCl3, 400 MHz): δ 0.80 (t, J = 7.5 Hz, 3H), 0.95 (d, J = 6.3 Hz, 3H), 1.00 (d, J = 6.3 Hz, 3H, CH3CH2), 1.19 (br s, 1H, NH), 1.51–1.60 (m, 1H, CH3CH2), 1.66–1.75 (m, 1H, CH3CH2), 2.62–2.78 (m, 3H, CH2NH and CH2CHCH2), 2.85–2.93 (m, 1H, NHCH), 7.14–7.25 (m, 3H, Ar–H), 7.27–7.33 (m, 2H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 12.0, 22.7, 22.8, 27.6, 47.9, 48.5, 53.0, 126.3, 127.8 (2C), 128.4 (2C), 143.8 ppm; HRMS (ESI, m/z) calcd for [C13H22N]+ (M + H+): 192.1752; found: 192.1750.
:3), the known amine18e2r (137 mg, yield: 65%) as a pale yellow oil. IR (film) νmax: 3332, 3029, 2959, 2926, 2843, 1603, 1503, 1449, 1358, 761, 699 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.37 (dd, J = 6.6, 0.8 Hz, 3H, CHCH3), 1.71 (br s, 1H, NH), 3.59 (d, J = 13.1 Hz, 1H, CH2NH), 3.66 (d, J = 13.1 Hz, 1H, CH2NH), 3.81 (q, J = 6.7 Hz, 1H, NHCH), 7.20–7.40 (m, 10H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 24.5, 51.6, 57.5, 126.7 (2C), 126.8, 126.9, 128.1 (2C), 128.3 (2C), 128.4 (2C), 140.6, 145.5 ppm; MS (ESI, m/z): 212 (M + H+).
:3), the known amine18f2s (116 mg, yield: 71%) as a pale yellow oil. IR (film) νmax: 3316, 3063, 2955, 2918, 2851, 1603, 1499, 1462, 1130, 740, 694 cm−1; 1H NMR (CDCl3, 400 MHz): δ 0.91 (t, J = 7.3 Hz, 3H, CH2CH3), 1.30–1.40 (m, 2H, CH2CH2CH3), 1.43 (br s, 1H, NH), 1.46–1.54 (m, 2H, CH2CH2CH3), 2.63 (t, J = 7.3 Hz, 2H, NHCH2CH2), 3.78 (s, 2H, CH2NH), 7.21–7.28 (m, 1H, Ar–H), 7.28–7.34 (m, 4H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 14.0, 20.4, 32.2, 49.2, 54.1, 126.8, 128.1 (2C), 128.3 (2C), 140.5 ppm; MS (ESI, m/z): 164 (M + H+).
:3), the known amine12d2t (153 mg, yield: 72%) as a pale yellow oil. IR (film) νmax: 3324, 3063, 3021, 2926, 2851, 2818, 1599, 1503, 1449, 1122, 1035, 753, 694 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.55 (br s, 1H, NH), 2.80–2.86 (m, 2H, NHCH2CH2), 2.88–2.93 (m, 2H, NHCH2CH2), 3.80 (s, 2H, CH2NH), 7.17–7.25 (m, 4H, Ar–H), 7.25–7.33 (m, 6H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 36.3, 50.5, 53.8, 126.1, 126.9, 128.0 (2C), 128.3 (2C), 128.4 (2C), 128.7 (2C), 140.0, 140.2 ppm; MS (ESI, m/z): 212 (M + H+).
:3), the known amine18d2u (192 mg, yield: 74%) as a colourless oil. IR (film) νmax: 3320, 2934, 2851, 1487, 1458, 1122, 732, 693 cm−1; 1H NMR (CDCl3, 400 MHz): δ 0.88 (t, J = 7.0 Hz, 3H, CH2CH3), 1.23–1.32 (m, 16H, CH2(CH2)8CH2), 1.42 (br s, 1H, NH), 1.47–1.54 (m, 2H, CH2CH2CH3), 2.62 (t, J = 7.3 Hz, 2H, NHCH2CH2), 3.78 (s, 2H, NHCH2), 7.22–7.27 (m, 1H, Ar–H), 7.32 (m, 4H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 14.1, 22.7, 27.4, 29.3, 29.6 (4C), 30.1, 31.9, 49.5, 54.1, 126.8, 128.1 (2C), 128.3 (2C), 140.6 ppm; MS (ESI, m/z): 262 (M + H+).
:3), the known amine12e2v (66 mg, yield: 65%) as a pale yellow oil. IR (film) νmax: 3320, 3026, 2919, 2849, 1453, 1263, 1107, 735, 699 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.76 (br s, 1H, NH), 3.83 (s, 2H, NHCH2), 3.99 (s, 2H, CH2NH), 6.87–7.00 (m, 2H, Ar–H), 7.20–7.28 (m, 2H, Ar–H), 7.29–7.39 (m, 4H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 47.5, 52.7, 124.3, 124.8, 126.6, 127.0, 128.2 (2C), 128.4 (2C), 140.0, 144.2 ppm; MS (ESI, m/z): 204 (M + H+).
:3), amine 2w (87 mg, yield: 68%) as a pale yellow oil. IR (film) νmax: 3361, 3026, 2905, 2846, 1494, 1452, 1107, 733, 697 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.51–1.55 (m, 6H, CHCH2CH and NH), 1.61–1.67 (m, 4H, CHCH2CH), 1.69–1.74 (m, 3H, CHCH2CH), 1.97 (m, 3H, CH2CHCH2), 2.25 (s, 2H, CH2NH), 3.79 (s, 2H, NHCH2), 7.21–7.26 (m, 1H, Ar–H), 7.29–7.35 (m, 4H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 28.5 (3C), 33.5, 37.3 (3C), 40.9 (3C), 54.7, 62.2, 126.7, 127.9 (2C), 128.3 (2C), 141.0 ppm; HRMS (ESI, m/z) calcd for [C18H26N]+ (M + H+): 256.2065; found: 256.2061.
:3), the known amine12e2x (68 mg, yield: 56%) as a pale yellow oil. IR (film) νmax: 3336, 3021, 2916, 2849, 1607, 1501, 1495, 1441, 1246, 807, 736, 698 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.64 (br s, 1H, NH), 3.71 (s, 2H, CH2NH), 3.78 (s, 2H, NHCH2), 5.92 (s, 2H, OCH2O), 6.73–6.80 (m, 2H, Ar–H), 6.86 (s, 1H, Ar–H), 7.23–7.28 (m, 1H, Ar–H), 7.29–7.37 (m, 4H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 52.9 (2C), 100.8, 108.0, 108.7, 121.2, 126.9, 128.1 (2C), 128.4 (2C), 134.3, 140.3, 146.5, 147.7 ppm; MS (ESI, m/z): 242 (M + H+).
:3), amine 6 (56 mg, yield: 64%) as a pale yellow oil. IR (film) νmax: 3312, 2971, 2934, 2855, 1462, 1453, 1375, 1179, 735 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.12 (d, J = 6.3 Hz, 6H, CHCH3), 1.24 (t, J = 7.6 Hz, 3H, CH2CH3), 1.36 (br s, 1H, NH), 2.71 (q, J = 7.6 Hz, 2H, CH2CH3), 2.89 (heptet, J = 6.3 Hz, 1H, NHCH), 3.77 (s, 2H, CH2NH), 7.14–7.22 (m, 3H, Ar–H), 7.30 (d, J = 7.1 Hz, 1H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 15.4, 23.0 (2C), 25.3, 48.7, 48.8, 125.9, 127.1, 128.5, 128.8, 138.0, 142.2 ppm; HRMS (ESI, m/z) calcd for [C12H20N]+ (M + H+): 178.1596; found: 178.1596.
:3), amine 8 (116 mg, yield: 60%) as a pale yellow oil. IR (film) νmax: 3324, 3054, 2965, 2929, 1610, 1588, 1470, 1375, 1259, 1168, 794, 702, 636 cm−1; 1H NMR (CDCl3, 400 MHz): δ 0.96 (d, J = 6.3 Hz, 6H, CHCH3), 1.36 (br s, 1H, NH), 2.67 (heptet, J = 6.3 Hz, 1H, NHCH), 3.64 (s, 2H, CH2NH), 7.10 (d, J = 8.1 Hz, 1H, Ar–H), 7.22–7.26 (m, 1H, Ar–H), 7.28–7.33 (m, 2H, Ar–H), 7.41–7.48 (m, 2H, Ar–H), 7.64 (m, 1H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 22.7 (2C), 48.4, 48.7, 119.2, 119.8, 121.6, 121.8, 122.2, 127.4, 129.6, 130.1, 131.4, 132.3, 139.0, 140.3, 142.1, 149.1 ppm; HRMS (ESI, m/z) calcd for [C17H18BrF3NO]+ (M + H+): 388.0524 and 390.0503; found: 388.0512 and 390.0503.
:3), amine 10 (96 mg, yield: 61%) as a pale yellow oil. IR (film) νmax: 3328, 3056, 2962, 2867, 1499, 1457, 1171, 760, 702 cm−1; 1H NMR (CDCl3, 400 MHz): δ 0.69 (d, J = 6.3 Hz, 6H, CHCH3), 0.91 (br s, 1H, NH), 2.28–2.40 (m, 3H, CH2CH2 and NH), 2.69–2.76 (m, 2H, CH2CH2NH), 7.16–7.22 (m, 2H, Ar–H), 7.23–7.29 (m, 1H, Ar–H), 7.31–7.44 (m, 10H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 22.6 (2C), 30.9, 47.1, 47.2, 125.3, 126.9 (2C), 128.1 (4C), 129.2 (4C), 129.4 (2C), 135.3 (2C), 142.2 (2C), 142.9 ppm; HRMS (ESI, m/z) calcd for [C23H26N]+ (M + H+): 316.2065; found: 316.2062.
:5), pyrrolidine 12 (80 mg, yield: 82%) as a colourless oil. [α]20D +152.7 (c 1.0, CHCl3); IR (film) νmax: 3440, 3034, 2926, 2872, 1495, 1453, 1180, 1047, 740, 707 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.84–1.93 (m, 1H, OCHCH2CH2), 2.00–2.09 (m, 1H, OCHCH2CH2), 2.87–3.14 (m, 3H, NHCH2 and PhCH2CH), 3.38–3.46 (dd, J = 12.1, 3.1 Hz, 1H, PhCH2CH), 3.46–3.56 (m, 1H, NHCH), 3.79 (t, J = 3.4 Hz, 1H, OCHCH2), 3.98 (br s, 1H, NH), 4.30 (d, J = 11.3 Hz, 1H, OCH2Ph), 4.50 (d, J = 11.3 Hz, 1H, OCH2Ph), 7.16–7.43 (m, 10H, Ar–H) ppm; 13C NMR (CDCl3, 100 MHz): δ 27.7, 31.9, 53.0, 70.6, 72.3, 77.7, 126.6, 127.8, 128.0, 128.6 (2C), 129.1, 137.4, 137.9 ppm; HRMS (ESI, m/z) calcd for [C18H21NNaO]+ (M + Na+): 290.1521; found: 290.1510.
:20), the known proline derivative18g,h14 (75 mg, yield: 80%) as a white solid. Mp: 88–89 °C (EtOAc–hexane); [α]20D −47.6 (c 1.0, CHCl3 {lit.18i [α]24D −40.5 (c 0.4, MeOH)}. IR (film) νmax: 3443, 2980, 2930, 1727, 1379, 1180, 1159 cm−1; 1H NMR (CDCl3, 400 MHz): δ 1.49 (s, 9H, CH3), 1.75–1.83 (m, 1H, CH2CH2CH2), 1.89–1.98 (m, 1H, CH2CH2CH2), 2.00–2.11 (m, 1H, CH2CH2CH), 2.29–2.41 (m, 1H, CH2CH2CH), 2.87–2.98 (m, 1H, CH2CH2NH), 3.33–3.42 (m, 1H, CH2CH2NH), 3.63–3.71 (m, 1H, COCHNH), 4.98 (br s, 1H, NH) ppm; 13C NMR (CDCl3, 100 MHz): δ 24.8, 27.9 (3C), 30.0, 54.9, 67.3, 83.6, 171.2 ppm; HRMS (ESI, m/z) calcd for [C9H17NNaO2]+ (M + Na+): 194.1157; found: 194.1150.
Acknowledgements
The authors are grateful to the Natural Science Foundation of China (NSFC, grant no. 21332007), and the Program for Changjiang Scholars and Innovative Research Team in the University of the Ministry of Education of China for financial support.Notes and references
- For related reviews, see: (a) J. Magano and J. R. Dunetz, Org. Process Res. Dev., 2012, 16, 1156 CrossRef CAS; (b) M. A. Ricci, Modern Amination Methods, Wiley, New York, 2000; For selected recent examples, see: Search PubMed; (c) M. Mewald, J. W. Medley and M. Movassaghi, Angew. Chem., Int. Ed., 2014, 53, 11634 CrossRef CAS PubMed; (d) S. Bonazzi, B.-C. Cheng, J. S. Wzorek and D. A. Evans, J. Am. Chem. Soc., 2013, 135, 9338 CrossRef CAS PubMed; (e) Y. Tao, D. W. Widlicka, P. D. Hill, M. Couturier and G. R. Young, Org. Process Res. Dev., 2012, 16, 1805 CrossRef CAS; (f) Z.-H. Chen, Z.-M. Chen, Y.-Q. Zhang, Y.-Q. Tu and F.-M. Zhang, J. Org. Chem., 2011, 76, 10173 CrossRef CAS PubMed; (g) T. Taniguchi and H. Ishibashi, Tetrahedron, 2008, 64, 8773 CrossRef CAS PubMed; (h) A. Deiters, M. Pettersson and S. F. Martin, J. Org. Chem., 2006, 71, 6547 CrossRef CAS PubMed; (i) A. Padwa and J. D. Ginn, J. Org. Chem., 2005, 70, 5197 CrossRef CAS PubMed; (j) Y. Choi, H. Ishikawa, J. Velcicky, G. I. Elliott, M. M. Miller and D. L. Boger, Org. Lett., 2005, 7, 4539 CrossRef CAS PubMed; (k) P. Magnus, L. Gazzard, L. Hobson, A. H. Payne, T. J. Rainey, N. Westlund and V. Lynch, Tetrahedron, 2002, 58, 3423 CrossRef CAS; (l) Y. Nagao, W. M. Dai, M. Ochiai, S. Tsukagoshi and E. Fujita, J. Org. Chem., 1990, 55, 1148 CrossRef CAS.
- J. Seyden-Penne, Reductions by Alumino and Borohydrides in Organic Synthesis, Wiley, New York, 2nd edn, 1997 Search PubMed.
- (a) F. Louafi, J. Moreau, S. Shahane, S. Golhen, T. Roisnel, S. Sinbandhit and J. P. Hurvois, J. Org. Chem., 2011, 76, 9720 CrossRef CAS PubMed; (b) P.-Q. Huang, B.-G. Wei and Y.-P. Ruan, Synlett, 2003, 1663 CrossRef CAS PubMed; (c) J. C. McWilliams and J. Clardy, J. Am. Chem. Soc., 1994, 116, 8378 CrossRef CAS; (d) B. Rajashekhar and E. T. Kaiser, J. Org. Chem., 1985, 50, 5480 CrossRef CAS.
- (a) H. C. Brown and P. Heim, J. Org. Chem., 1973, 38, 912 CrossRef CAS; (b) S. Krishnamurthy, Tetrahedron Lett., 1982, 23, 3315 CrossRef CAS; (c) R. P. Polniaszek and C. R. Kaufman, J. Am. Chem. Soc., 1989, 111, 4859 CrossRef CAS; (d) M. Bonnat, A. Hercouet and M. Le Corre, Synth. Commun., 1991, 21, 1579 CrossRef CAS; see also: (e) A. S. B. Prasad, J. V. B. Kanth and M. Periasamy, Tetrahedron, 1992, 48, 4623 CrossRef CAS; (f) A. Giannis and K. Sandhoff, Angew. Chem., Int. Ed. Engl., 1989, 28, 218 CrossRef; (g) P.-Q. Huang, G. Chen and X. Zheng, J. Heterocycl. Chem., 2007, 44, 499 CrossRef CAS.
- H. C. Brown, S. Narasimhan and Y. M. Choi, Synthesis, 1981, 441 CrossRef CAS.
- N. Yamazaki, M. Atobe and C. Kibayashi, Tetrahedron Lett., 2001, 42, 5029 CrossRef CAS.
- (a) S. J. Paikoff, T. E. Wilson, C. Y. Cho and P. G. Schultz, Tetrahedron Lett., 1996, 37, 5653 CrossRef CAS; (b) P. J. Brown, K. P. Hurley, L. W. Stuart and T. M. Willson, Synthesis, 1997, 778 CrossRef CAS PubMed; (c) A. Nefzi, J. M. Ostresh, J. P. Meyer and R. A. Houghten, Tetrahedron Lett., 1997, 38, 931 CrossRef CAS; (d) D. G. Hall, C. Laplante, S. Manku and J. Nagendran, J. Org. Chem., 1999, 64, 698 CrossRef CAS; (e) M. Couturier, J. L. Tucker, B. M. Andresen, P. l. Dubé and J. T. Negri, Org. Lett., 2001, 3, 465 CrossRef CAS; (f) S. Manku, C. Laplante, D. Kopac, T. y. Chan and D. G. Hall, J. Org. Chem., 2001, 66, 874 CrossRef CAS.
- (a) H. Akamatsu, S. Kusumoto and K. Fukase, Tetrahedron Lett., 2002, 43, 8867 CrossRef CAS; (b) B. Thiedemann, C. M. L. Schmitz and A. Staubitz, J. Org. Chem., 2014, 79, 10284 CrossRef CAS PubMed.
- (a) M. A. Mauragis, M. F. Lipton and M. F. Veley, Org. Process Res. Dev., 2002, 6, 192 CrossRef CAS; (b) Stepwise: B. H. Lee and M. F. Clothier, Tetrahedron Lett., 1999, 40, 643 CrossRef CAS.
- For recent reviews, see: (a) A. M. Smith and R. Whyman, Chem. Rev., 2014, 114, 5477 CrossRef CAS PubMed; (b) S. Werkmeister, K. Junge and M. Beller, Org. Process Res. Dev., 2014, 18, 289 CrossRef CAS; (c) P. A. Dub and T. Ikariya, ACS Catal., 2012, 2, 1718 CrossRef CAS; (d) D. Addis, S. Das, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 6004 CrossRef CAS PubMed; (e) S. Das, S. Zhou, D. Addis, K. Junge, S. Enthaler and M. Beller, Top. Catal., 2010, 53, 979 CrossRef CAS; (f) Modern Reduction Methods, ed. P. G. Andersson and I. J. Munslow, Wiley, New York, 2008; For selected examples on the reduction of tertiary amides, see: Search PubMed; (g) M. Igarashi and T. Fuchikami, Tetrahedron Lett., 2001, 42, 1945 CrossRef CAS; (h) N. Sakai, K. Fujii and T. Konakahara, Tetrahedron Lett., 2008, 49, 6873 CrossRef CAS PubMed; (i) G. Barbe and A. B. Charette, J. Am. Chem. Soc., 2008, 130, 18 CrossRef CAS PubMed; (j) S.-L. Zhou, K. Junge, D. Addis, S. Das and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 9507 CrossRef CAS PubMed; (k) Y. Sunada, H. Kawakami, T. Imaoka, Y. Motoyama and H. Nagashima, Angew. Chem., Int. Ed., 2009, 48, 9511 CrossRef CAS PubMed; (l) S. Hanada, E. Tsutsumi, Y. Motoyama and H. Nagashima, J. Am. Chem. Soc., 2009, 131, 15032 CrossRef CAS PubMed; (m) S. Das, D. Addis, S.-L. Zhou, K. Junge and M. Beller, J. Am. Chem. Soc., 2010, 132, 1770 CrossRef CAS PubMed (correction: J. Am. Chem. Soc., 2010, 132, 4971); (n) S.-H. Xiang, J. Xu, H.-Q. Yuan and P.-Q. Huang, Synlett, 2010, 1829 CAS; (o) M. Stein and B. Breit, Angew. Chem., Int. Ed., 2013, 52, 2231 CrossRef CAS PubMed; (p) W.-L. Xie, M.-D. Zhao and C.-M. Cui, Organometallics, 2013, 32, 7440 CrossRef CAS; (q) E. Blondiaux and T. Cantat, Chem. Commun., 2014, 50, 9349 RSC; (r) J. D. Kirkham, R. J. Butlin and J. P. Harrity, Angew. Chem., Int. Ed., 2012, 51, 6402 CrossRef CAS PubMed.
- For selected recent examples on the reduction of secondary amides, see: (a) Y. Motoyama, K. Mitsui, T. Ishida and H. Nagashima, J. Am. Chem. Soc., 2005, 127, 13150 CrossRef CAS PubMed; (b) T. Ohta, M. Kamiya, M. Nobutomo, K. Kusui and I. Furukawa, Bull. Chem. Soc. Jpn., 2005, 78, 1856 CrossRef CAS; (c) A. C. Fernandes and C. C. Romão, J. Mol. Catal. A: Chem., 2007, 272, 60 CrossRef CAS PubMed; (d) A. A. Núñez Magro, G. R. Eastham and D. J. Cole-Hamilton, Chem. Commun., 2007, 3154 RSC; (e) S. Hanada, T. Ishida, Y. Motoyama and H. Nagashima, J. Org. Chem., 2007, 72, 7551 CrossRef CAS PubMed; For two early examples, see: (f) Atta-ur-Rahman, A. Basha, N. Waheed and S. Ahmed, Tetrahedron Lett., 1976, 17, 219 CrossRef; (g) R. F. Borch, Tetrahedron Lett., 1968, 9, 61 CrossRef.
- (a) E. A. Voight, M. S. Bodenstein, N. Ikemoto and M. H. Kress, Tetrahedron Lett., 2006, 47, 1717 CrossRef CAS PubMed; (b) G. Pelletier, W. S. Bechara and A. B. Charette, J. Am. Chem. Soc., 2010, 132, 12817 CrossRef CAS PubMed; (c) S. Das, D. Addis, K. Junge and M. Beller, Chem. – Eur. J., 2011, 17, 12186 CrossRef CAS PubMed; (d) S. Das, B. Join, K. Junge and M. Beller, Chem. Commun., 2012, 48, 2683 RSC; (e) C. Cheng and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 11304 CrossRef CAS PubMed; (f) J. T. Reeves, Z. Tan, M. A. Marsini, Z. S. Han, Y. Xu, D. C. Reeves, H. Lee, B. Z. Lu and C. H. Senanayake, Adv. Synth. Catal., 2013, 355, 47 CrossRef CAS; (g) T. Zhang, Y. Zhang, W. Zhang and M. Luo, Adv. Synth. Catal., 2013, 355, 2775 CrossRef CAS; (h) B. Ravinder, S. R. Reddy, A. P. Reddy and R. Bandichhor, Tetrahedron Lett., 2013, 54, 4908 CrossRef CAS PubMed; (i) J. A. M. e L. Torre, K. Grabow, U. Bentrup, K. Junge, S. Zhou, A. Brückner and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 11577 CrossRef PubMed.
- For a review on the use of a secondary amide as a directing group for lithiation, see: (a) P. Beak, A. Basu, D. J. Gallagher, Y. S. Peak and S. Thayumanavan, Acc. Chem. Res., 1996, 29, 552 CrossRef CAS; for selected examples, see: (b) P. Beak and D. J. Kempf, J. Am. Chem. Soc., 1980, 102, 4550 CrossRef CAS; (c) P. Beak, D. J. Kempf and K. D. Wilson, J. Am. Chem. Soc., 1985, 107, 4745 CrossRef CAS; (d) D. A. Evans and G. C. Fu, J. Am. Chem. Soc., 1991, 113, 4042 CrossRef CAS; (e) G. P. Lutz, H. Du, D. J. Gallagher and P. Beak, J. Org. Chem., 1996, 61, 4542 CrossRef CAS PubMed; (f) D. J. Pippel, M. D. Curtis, H. Du and P. Beak, J. Org. Chem., 1998, 63, 2 CrossRef CAS; (g) J. M. Laumer, D. D. Kim and P. Beak, J. Org. Chem., 2002, 67, 6797 CrossRef CAS PubMed; (h) K. Smith, G. A. El-Hiti and M. B. Alshammari, J. Org. Chem., 2012, 77, 11210 CrossRef CAS PubMed.
- (a) Q. Chen, L. Ilies, N. Yoshikai and E. Nakamura, Org. Lett., 2011, 13, 3232 CrossRef CAS PubMed; (b) D. Shabashov and O. Daugulis, Org. Lett., 2006, 8, 4947 CrossRef CAS PubMed; (c) C. S. Yeung, X. Zhao, N. Borduas and V. M. Dong, Chem. Sci., 2010, 1, 331 RSC.
- For selected direct transformation of secondary amides, see: (a) N. J. Sisti, F. W. Fowler and D. S. Grierson, Synlett, 1991, 816 CrossRef CAS PubMed; (b) D. J. A. Schedler, A. G. Godfrey and B. Ganem, Tetrahedron Lett., 1993, 34, 5035 CrossRef CAS; (c) D. J. A. Schedler, J. Li and B. Ganem, J. Org. Chem., 1996, 61, 4115 CrossRef CAS PubMed; (d) Q. Xia and B. Ganem, Org. Lett., 2001, 3, 485 CrossRef CAS; (e) Q. Xia and B. Ganem, Tetrahedron Lett., 2002, 43, 1597 CrossRef CAS; (f) M. Movassaghi and M. D. Hill, J. Am. Chem. Soc., 2006, 128, 4592 CrossRef CAS PubMed; (g) J. T. Spletstoser, J. M. White, A. R. Tunoori and G. I. Georg, J. Am. Chem. Soc., 2007, 129, 3408 CrossRef CAS PubMed; (h) H.-B. Zhou, G.-S. Liu and Z.-J. Yao, J. Org. Chem., 2007, 72, 6270 CrossRef CAS PubMed; (i) S.-L. Cui, J. Wang and Y.-G. Wang, J. Am. Chem. Soc., 2008, 130, 13526 CrossRef CAS PubMed; (j) Y. Oda, T. Sato and N. Chida, Org. Lett., 2012, 14, 950 CrossRef CAS PubMed; (k) W. S. Bechara, G. Pelletier and A. B. Charette, Nat. Chem., 2012, 4, 228 CrossRef CAS PubMed; (l) K.-J. Xiao, A.-E Wang, Y.-H. Huang and P.-Q. Huang, Asian J. Org. Chem., 2012, 1, 130 CrossRef CAS; (m) K.-J. Xiao, Y.-H. Huang and P.-Q. Huang, Acta Chim. Sin., 2012, 70, 1917 CrossRef CAS; (n) K.-J. Xiao, A.-E Wang and P.-Q. Huang, Angew. Chem., Int. Ed., 2012, 51, 8314 CrossRef CAS PubMed; (o) Y. Gao, Z. Huang, R. Zhuang, J. Xu, P. Zhang, G. Tang and Y. Zhao, Org. Lett., 2013, 15, 4214 CrossRef CAS PubMed; (p) K. Shirokane, T. Wada, M. Yoritate, R. Minamikawa, N. Takayama, T. Sato and N. Chida, Angew. Chem., Int. Ed., 2014, 53, 512 CrossRef CAS PubMed; (q) M. Szostak, M. Spain, A. J. Eberhart and D. J. Procter, J. Am. Chem. Soc., 2014, 136, 2268 CrossRef CAS PubMed; (r) T. Sato and N. Chida, Org. Biomol. Chem., 2014, 12, 3147 RSC; (s) M. Yoritate, T. Meguro, N. Matsuo, K. Shirokane, T. Sato and N. Chida, Chem. – Eur. J., 2014, 20, 8210 CrossRef CAS PubMed; (t) M. Nakajima, Y. Oda, T. Wada, R. Minamikawa, K. Shirokane, T. Sato and N. Chida, Chem. – Eur. J., 2014, 20, 17565 CrossRef CAS PubMed.
- (a) K.-J. Xiao, J.-M. Luo, K.-Y. Ye, Y. Wang and P.-Q. Huang, Angew. Chem., Int. Ed., 2010, 49, 3037 CrossRef CAS PubMed; (b) K.-J. Xiao, Y. Wang, K.-Y. Ye and P.-Q. Huang, Chem. – Eur. J., 2010, 16, 12792 CrossRef CAS PubMed; (c) S.-Y. Huang, Z. Chang, S.-C. Tuo, L.-H. Gao, A.-E Wang and P.-Q. Huang, Chem. Commun., 2013, 49, 7088 RSC; (d) K.-J. Xiao, Y. Wang, Y.-H. Huang, X.-G. Wang, J.-C. Liao and P.-Q. Huang, J. Org. Chem., 2013, 78, 8305 CrossRef CAS PubMed; (e) K.-J. Xiao, J.-M. Luo, X.-E. Xia, Y. Wang and P.-Q. Huang, Chem. – Eur. J., 2013, 19, 13075 CrossRef CAS PubMed; (f) P.-Q. Huang, W. Ou, K.-J. Xiao and A.-E Wang, Chem. Commun., 2014, 50, 8761 RSC; (g) H.-Q. Deng, X.-Y. Qian, Y.-X. Li, J.-F. Zheng, L.-F. Xie and P.-Q. Huang, Org. Chem. Front., 2014, 1, 258 RSC.
- For reviews on the chemistry of triflic acid and its derivatives, see: (a) P. J. Stang, M. Hanack and M. L. R. Subramanian, Synthesis, 1983, 85 Search PubMed; (b) I. L. Baraznenok, V. G. Nenajdenko and E. S. Balenkova, Tetrahedron, 2000, 56, 3077 CrossRef CAS; For selected recent examples on the use of Tf2O as a powerful amide activator for a variety of transformations, see: (c) G. Bélanger, R. Larouche-Gauthier, F. Ménard, M. Nantel and F. Barabé, J. Org. Chem., 2006, 71, 704 CrossRef PubMed; (d) B. Peng, X. Huang, L.-G. Xie and N. Maulide, Angew. Chem., Int. Ed., 2014, 53, 8718 CrossRef CAS PubMed; (e) B. Peng, D. Geerdink, C. Farès and N. Maulide, Angew. Chem., Int. Ed., 2014, 53, 5462 CrossRef CAS PubMed.
- (a) O. Saidi, A. J. Blacker, M. M. Farah, S. P. Marsden and J. M. J. Williams, Angew. Chem., Int. Ed., 2009, 48, 7375 CrossRef CAS PubMed; (b) S. W. C. Bornschein, K. Junge and M. Beller, Eur. J. Org. Chem., 2013, 3671 Search PubMed; (c) N. Fleury-Brgeot, J. Raushel, D. L. Sandrock, S. D. Dreher and G. A. Molander, Chem. – Eur. J., 2012, 18, 9564 CrossRef PubMed; (d) R. A. Sanchez-Delgado, R. G. D. Rosa and E. Ocando-Mavarezc, J. Mol. Catal. A: Chem., 1996, 108, 125 CrossRef CAS; (e) C. Guérin, V. Bellosta, G. Guillamot and J. Cossy, Org. Lett., 2011, 13, 3534 CrossRef PubMed; (f) W. Zhang, X. Dong and W. Zhao, Org. Lett., 2011, 13, 5386 CrossRef CAS PubMed; (g) A. H. G. Siebum, R. K. F. Tsang, R. van der Steen, J. Raap and J. Lugtenburg, Eur. J. Org. Chem., 2004, 4391 CrossRef CAS; (h) S. Tanaka, H. Saburi, T. Murase, M. Yoshimura and M. Kitamura, J. Org. Chem., 2006, 71, 4682 CrossRef CAS PubMed; (i) G. Jolles, G. Poiget, J. Robert, B. Terlain and J. P. Thomas, Bull. Soc. Chim. Fr., 1965, 8, 2252 Search PubMed.
- S.-H. Xiang, H.-Q. Yuan and P.-Q. Huang, Tetrahedron: Asymmetry, 2009, 20, 2021 CrossRef CAS PubMed.
- (a) J. Schwartz and J. A. Labinger, Angew. Chem., Int. Ed. Engl., 1976, 15, 333 CrossRef; (b) B. Ganem and R. R. Franke, J. Org. Chem., 2007, 72, 3981 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of all reduction products. See DOI: 10.1039/c4qo00317a |
| This journal is © the Partner Organisations 2015 |