DOI:
10.1039/C4QO00310A
(Research Article)
Org. Chem. Front., 2015,
2, 159-162
Hg(OAc)2 mediated highly regio- and/or diastereoselective allylic tert-acetylation of olefins†
Received
25th November 2014
, Accepted 26th December 2014
First published on 30th December 2014
Abstract
Isolation of reaction intermediates facilitated a mechanistic investigation of the known Hg(OAc)2 mediated aromatisation reaction of carvone. We propose a different mechanism for this reaction than the previously proposed mechanism. The reaction method was further exploited for the highly regio- and/or diastereoselective allylic oxidation of geminally disubstituted olefins to allylic tert-acetates and applied in the total synthesis of andirolactone.
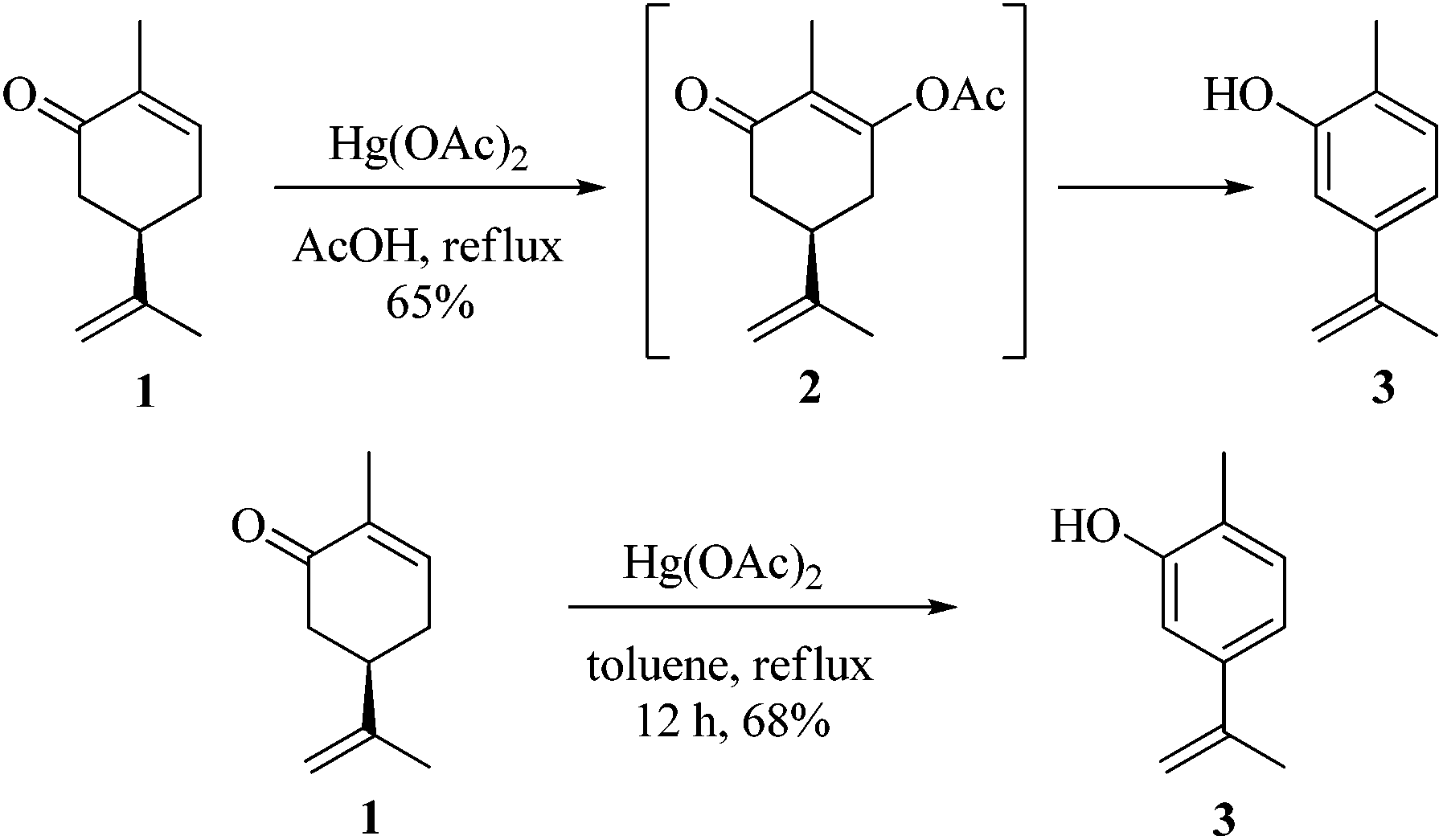
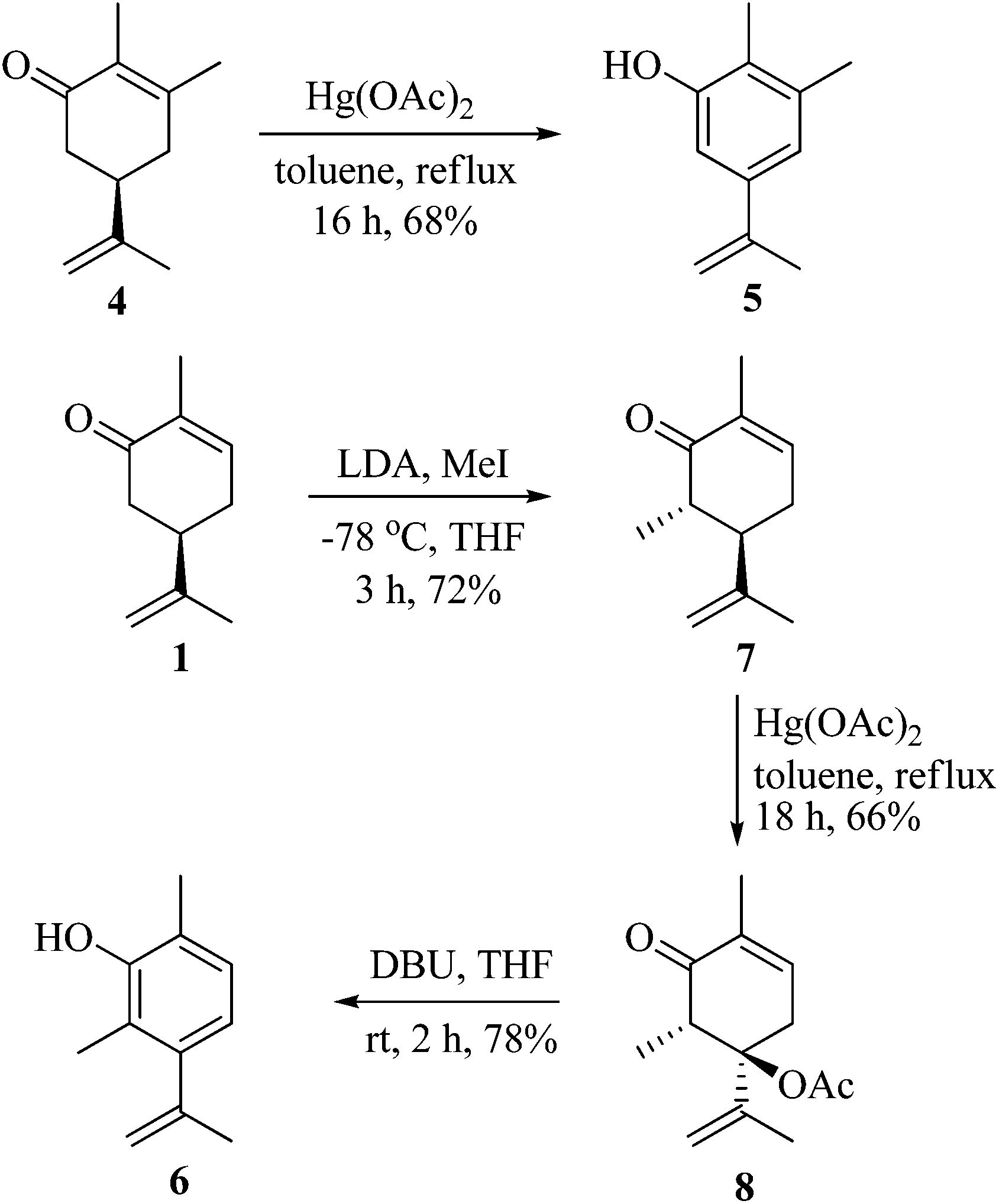
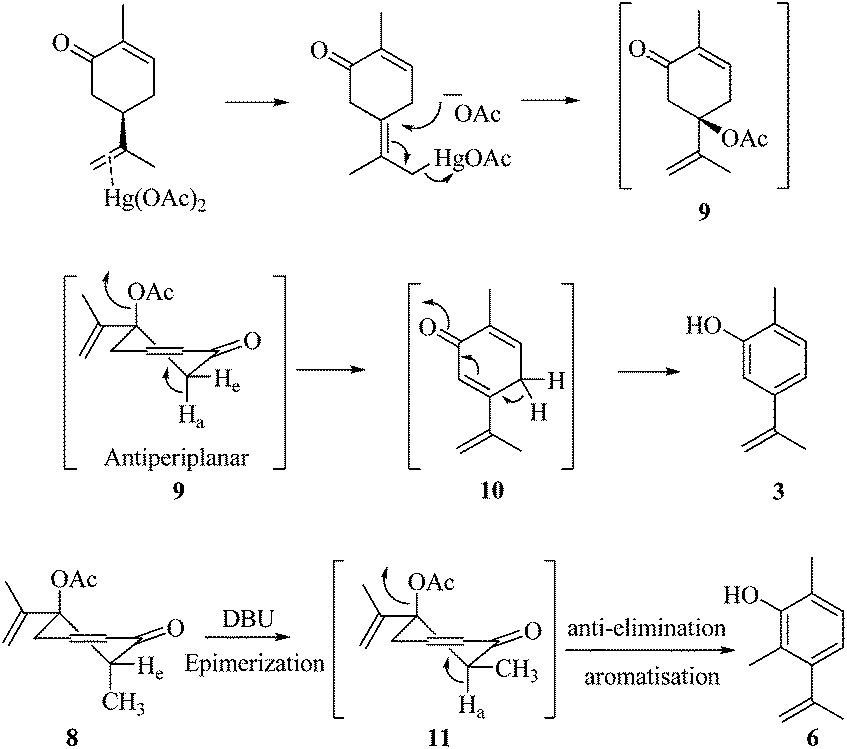
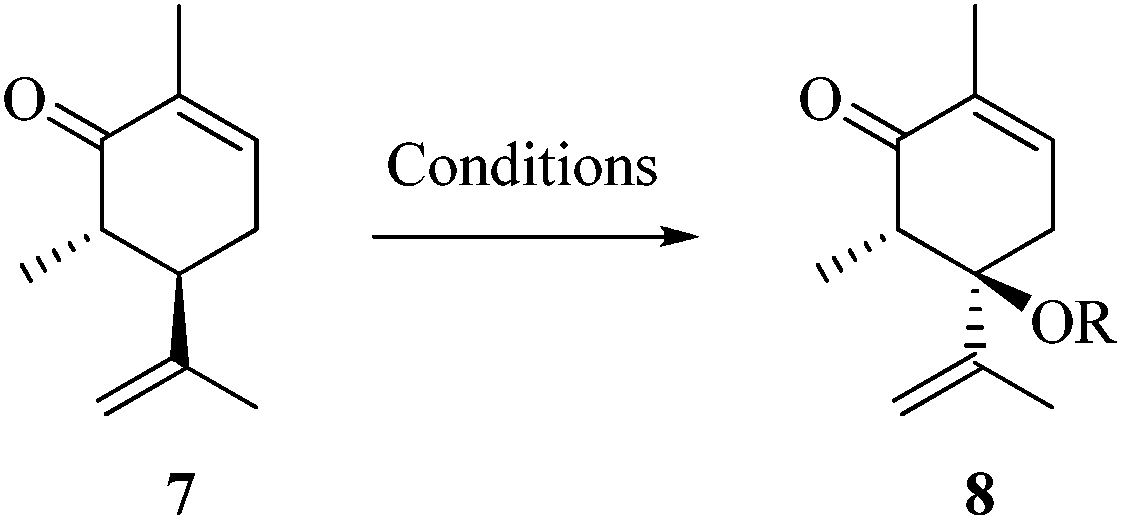
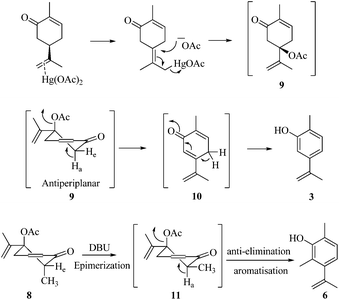
As part of our ongoing research towards the development of novel reactions and methods for the synthesis of highly substituted indenes,1 we came across a literature report, where Treibs2 had achieved the aromatisation of carvone 1 using a stoichiometric amount of Hg(OAc)2. We thought of exploiting this reaction for the synthesis of substituted aromatic derivatives which are otherwise not trivial to make by conventional methods.3 In the original report, Treibs2 reported the aromatisation of carvone 1 using a stoichiometric amount of Hg(OAc)2 in acetic acid under reflux conditions. Treibs proposed that when treated with Hg(OAc)2 under acidic conditions, carvone 1 might be forming intermediate 2 which upon further elimination forms the phenol derivative 3 (Scheme 1). After screening different solvents and temperatures, we found that carvone 1 could be aromatised in 68% yield using a stoichiometric amount of Hg(OAc)2 in toluene under reflux conditions without using AcOH or any other acid.4 After standardisation of the reaction, we next turned our attention towards the mechanism of the reaction (Scheme 2). As Treibs proposed that compound 2 could be the intermediate in the aromatisation reaction, it was thought that substitution at the 3-position of carvone 1 might prevent its aromatisation as per the proposed mechanism. So to validate the proposed mechanism, we treated 3-methylcarvone (4) with Hg(OAc)2 in toluene under reflux conditions. Surprisingly, under these reaction conditions, compound 4 aromatised to furnish compound 5 in 68% yield, suggesting that the proposed mechanism might not be correct. All of our efforts to isolate the intermediate of the carvone aromatisation reaction failed but interestingly, in an attempt to make the substituted aromatic compound 6, when we treated trans-6-methylcarvone (7) with Hg(OAc)2 in toluene under reflux conditions, instead of isolating the corresponding aromatic compound 6 we isolated compound 8, which has an acetate group at the C-5 position of methylcarvone 7, as the only diastereomer in 66% yield. The structure of compound 8 was established from spectroscopic analysis (IR, 1H, 13C NMR and HRMS). Treatment of compound 8 with a catalytic amount of DBU in CH2Cl2 furnished the aromatised product 6 in 78% yield presumably via epimerization, further confirming the structure of compound 8. The stability of compound 8 under these reaction conditions could be explained by syn and anti elimination chemistry. Upon treatment with Hg(OAc)2, carvone 1 generates intermediate 9in situ, which then undergoes facile anti-elimination, since one of the hydrogens α to the keto group is antiperiplanar to the acetate group, to give dienone 10 under the reaction conditions. Further isomerisation of dienone 10 yields phenol 3. But when reacted with Hg(OAc)2, trans-6-methylcarvone 7 forms the tertiary acetate 8 as shown in Scheme 3. The difference between intermediates 9 and 8 is that, in the case of 9, there is a hydrogen antiperiplanar to the acetate group which undergoes facile E2 elimination, so it is not possible to isolate intermediate 9. Since the tertiary acetate 8 does not have an acidic proton antiperiplanar to the acetate group, elimination does not take place and so in this case intermediate 8 is isolable unlike intermediate 9.
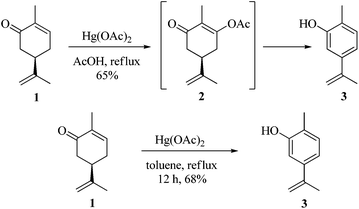 |
| | Scheme 1 Mercury acetate mediated aromatisation of carvone. | |
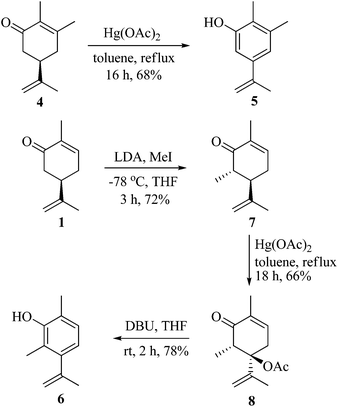 |
| | Scheme 2 Aromatisation of carvone derivatives. | |
 |
| | Scheme 3 Proposed mechanism for carvone aromatisation. | |
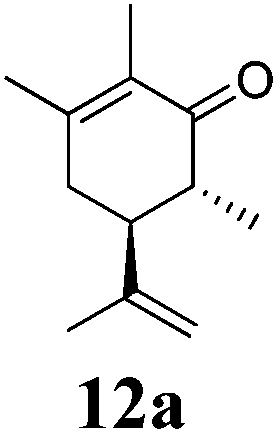
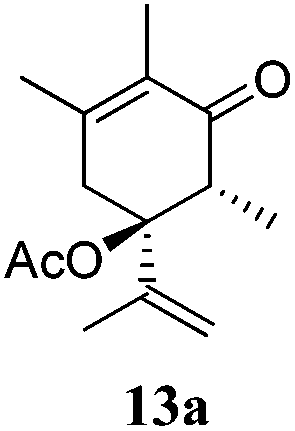
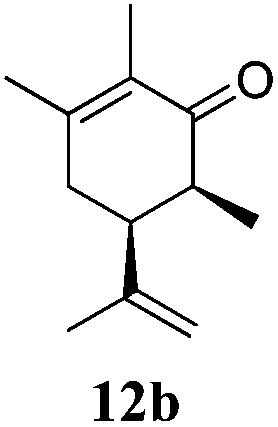
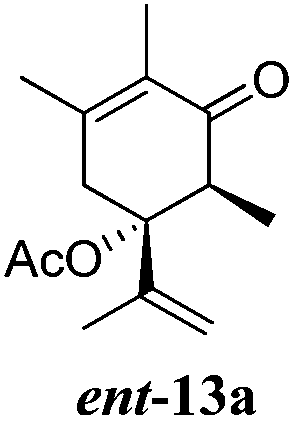
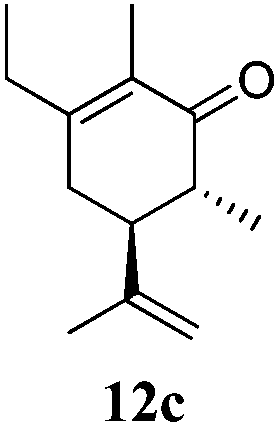
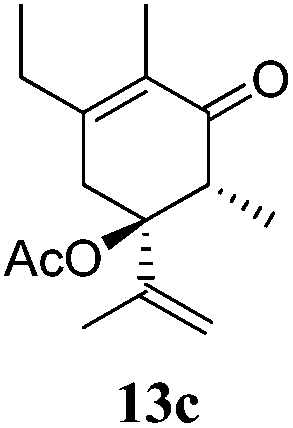
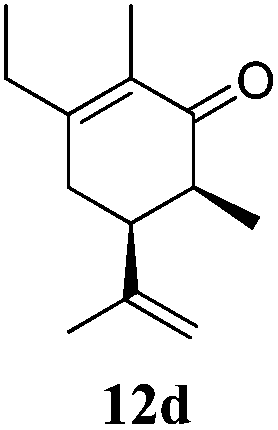
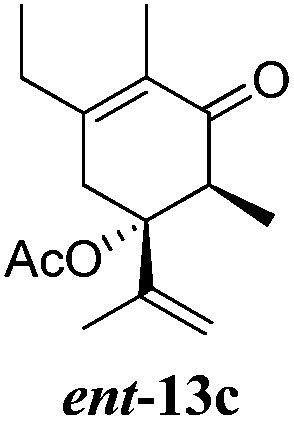
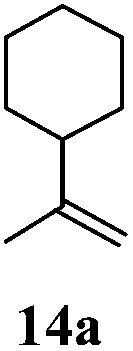
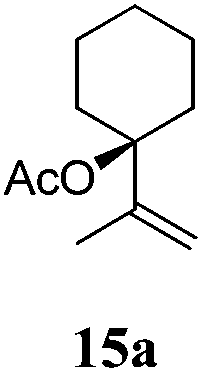
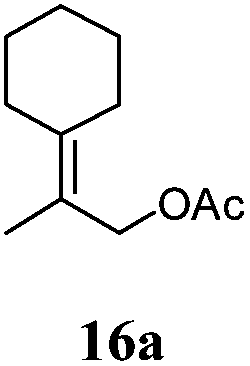
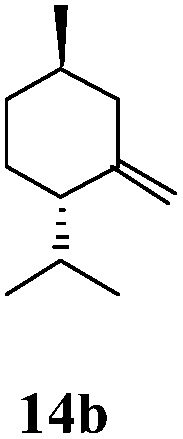
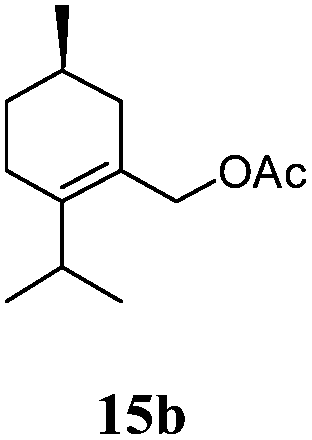
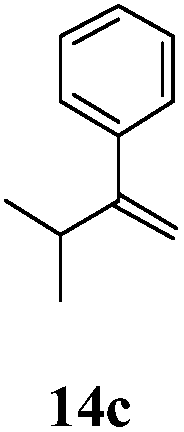
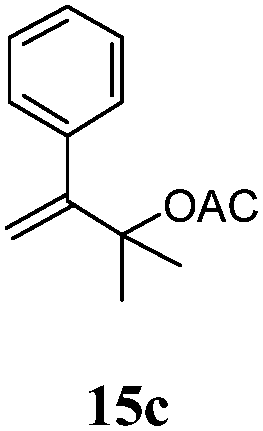
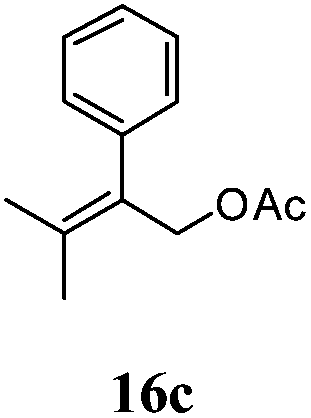
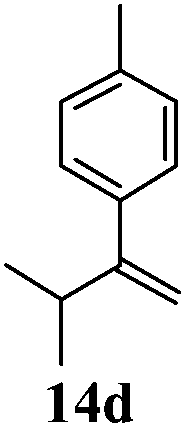
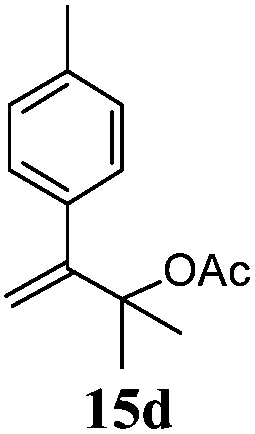
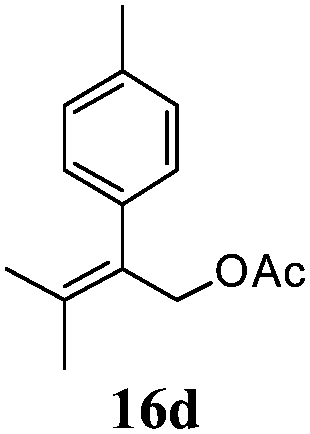
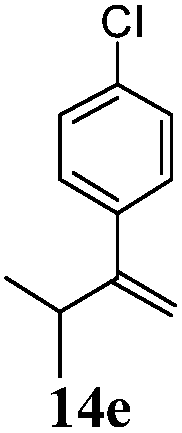
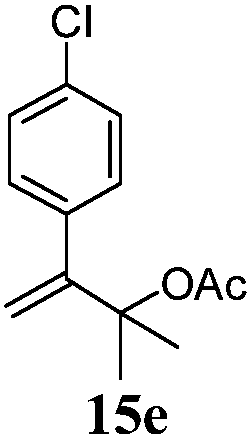
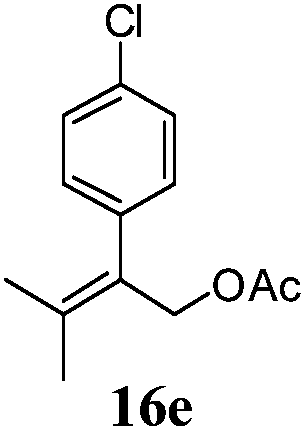
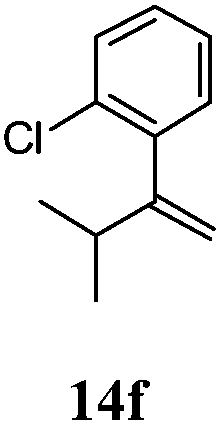
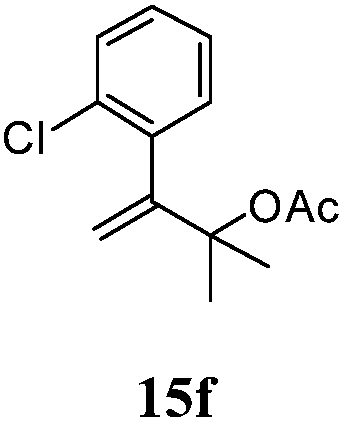
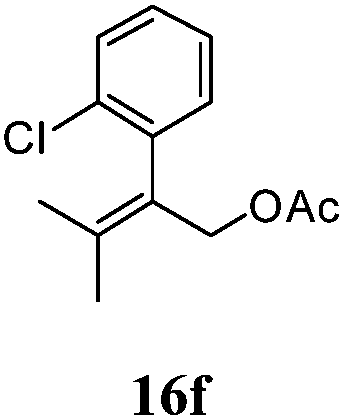
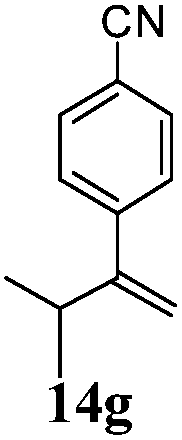
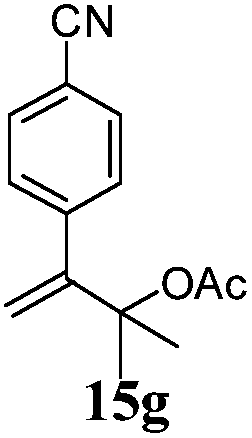
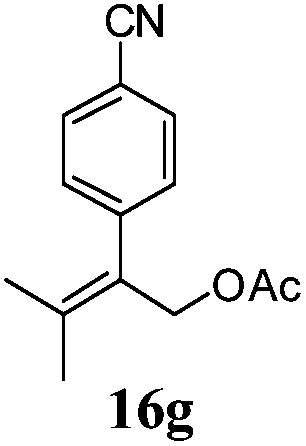
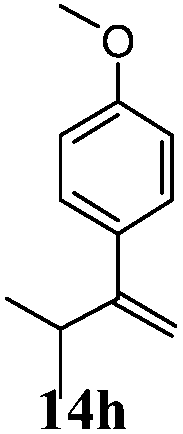
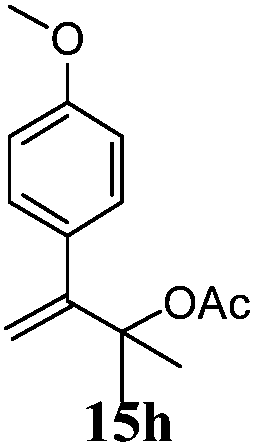
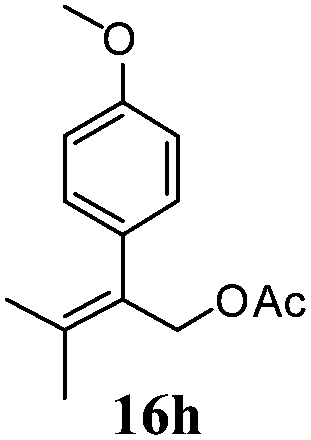
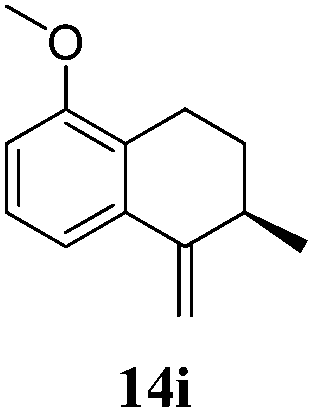
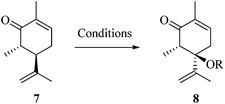
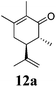
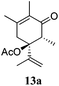
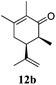
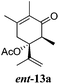
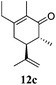
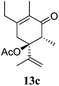
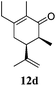
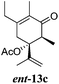
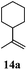
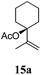
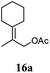
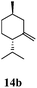
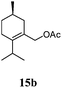
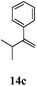
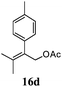
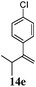
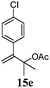
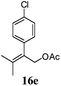
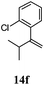
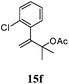
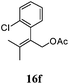
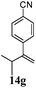
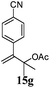
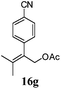
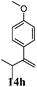
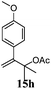
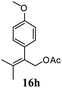
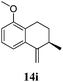
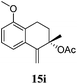
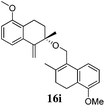
After establishing the mechanism of the aromatisation reaction and impressed by the diastereoselectivity and regioselectivity of this reaction, we thought of exploiting this reaction for the allylic acetylation of olefins. Although allylic oxidation of olefins by mercury acetate is well documented in literature,5 due to poor selectivity, yields and the toxicity of mercury, it was later replaced by more efficient catalytic methods using palladium based catalysts. We also screened different palladium,6 copper7 and selenium8 catalysts for the same transformation, but unfortunately did not generate any allylic oxidation products, instead in most of the cases we observed very poor yields with products that had multiple tlc spots or recovered the starting material in some cases (Table 1). Considering the fact that currently there is no method available in the literature for such a selective allylic tert-acetylation, we thought of testing the generality and substrate scope of this reaction. To our delight, the reaction showed broader substrate scope and generated the tert-acetylation product in a highly regioselective manner (Table 2). When independently treated with Hg(OAc)2 in refluxing toluene, the diastereomers 12a and 12b furnished compound 13a and its optical antipode ent-13a in excellent diastereoselectivity. In both 12a and 12b the approach of the incoming acetate group is controlled by the adjacent methyl group, hence in 12a and 12b, it generates trans isomers with respect to the acetate and the chiral methyl group. Thus, we were able to a generate pair of enantiomers (13a and ent-13a) from two diastereomers (12a & 12b). In the case of menthene 14b, we were not able to isolate the expected tert-acetate compound, instead it furnished the allylic secondary acetate 15b as the only product in 71% yield. We also explored various styrene derivatives for the allylic acetylation reaction (Table 3). All styrene derivatives generated the expected acetylation product in moderate yield (56–74%) along with rearranged secondary acetates (6–12%). Tetralin and indene derivatives (entry 14i and 14j) afforded acetates 15i and 15j in 60% and 59% yield respectively along with a minor amount of the dimers 16i and 16j.
Table 1 Screening of catalysts for the oxidation of methyl carvone 7 to 8
|
|
| Entry |
Conditions |
R |
Yielda (%) |
|
Isolated yield.
Decomposition of starting material observed.
Recovered starting material.
|
| 1 |
Pd(OAc)2, BQ, DMSO, AcOH |
–Ac |
Nilb |
| 2 |
Pd(TFA)2, 2-methoxyacetophenone, BQ, AcOH |
–Ac |
Nilb |
| 3 |
CuBr, PhCO3tBu, toluene |
–Bz |
Nilc |
| 4 |
Cu(OAc)2, PhCO3tBu, toluene |
–Bz |
Nilb |
| 5 |
CuCl, PhCO3tBu, toluene |
–Bz |
Nilc |
| 6 |
SeO2, tBuOOH, DCM |
-H |
6 |
| 7 |
SeO2, H2O2 |
-H |
8 |
| 8 |
Hg(OAc)2, toluene |
–Ac |
66 |
Table 2 Regioselective allylic acetylation of optically active cyclohexene derivativesa
| Entry |
Substrate |
Product |
de (%) |
Yield (%) |
|
Isolated yields, 1.2 eq. of mercury acetate was used.
|
| 1 |
|
|
>99 |
64 |
| 2 |
|
|
>99 |
66 |
| 3 |
|
|
>99 |
64 |
| 4 |
|
|
>99 |
67 |
Table 3 Regioselective allylic oxidation of geminally disubstituted olefins to allylic tert-acetatesa
| Entry |
Substrate |
Product |
Ratio (A![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :B) :B) |
Yield (%) |
| Major (A) |
Minor (B) |
|
Isolated yields, 1.2 eq. of mercury acetate was used.
|
| 1 |
|
|
|
5:1 |
67 |
| 2 |
|
|
— |
— |
71 |
| 3 |
|
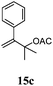
|
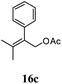
|
11:1 |
70 |
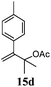
| 4 |
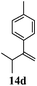
|
|
|
9:1 |
78 |
| 5 |
|
|
|
10:1 |
73 |
| 6 |
|
|
|
10:1 |
77 |
| 7 |
|
|
|
11:1 |
81 |
| 8 |
|
|
|
9:1 |
71 |
| 9 |
|
|
|
5:1 |
72 |
| 10 |
|
|
|
6:1 |
68 |
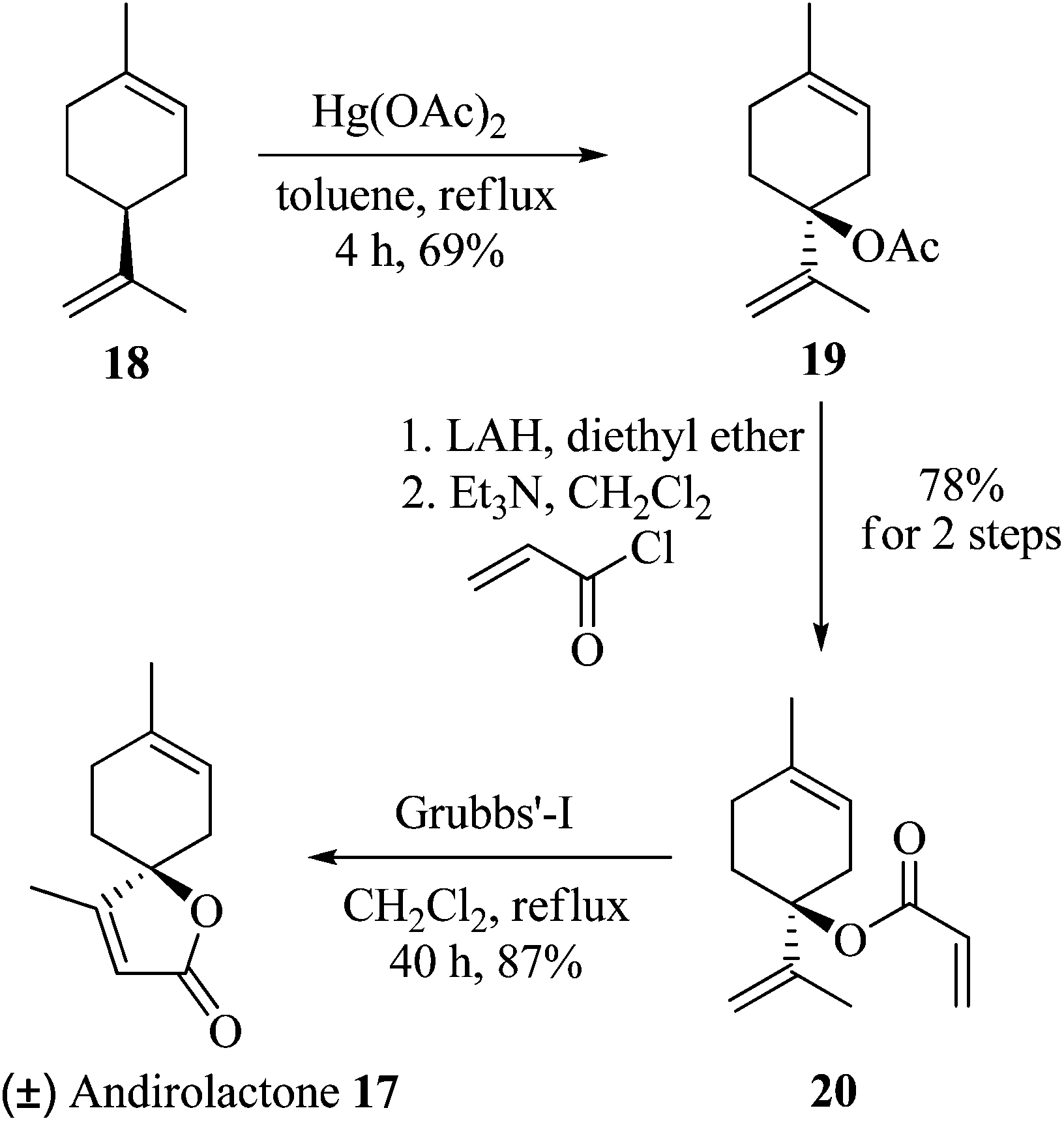
Finally, this reaction was applied for the total synthesis of the natural product andirolactone (17). In 1987, Avcibasi et al., isolated a spirocyclic butenolide terpenoid, andirolactone (17), with potential biological and medicinal properties, from the wood of Lebanese cedar (Cedrus libanotica).9 To date there have been 7 total syntheses of andirolactone reported in the literature.10 Herein, we report the concise synthesis of andirolactone. It was envisioned that andirolactone (17) could be synthesized from limonene 18 by its regioselective tert allylic oxidation, and reduction of the resulting ester to a tertiary alcohol, followed by treatment with acryloyl chloride and finally ring closing metathesis (RCM) of the diene thus generated. So limonene 18, having five allylic positions available for oxidation, when treated with Hg(OAc)2 in refluxing toluene generated tert acetate 19 in 69% yield (83% brsm) in a highly regioselective manner. Reduction of acetate 19 to a tertiary alcohol, followed by treatment with acryloyl chloride gave the diene intermediate 20. RCM of diene 20 using 5 mol% of Grubbs’ first generation catalyst afforded the natural product andirolactone (17) in 87% yield (46.8% overall yield) (Scheme 4).
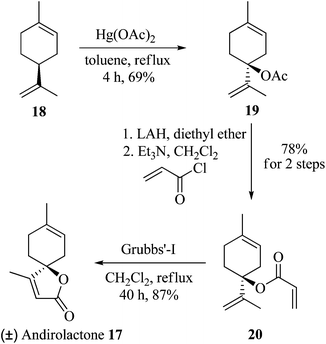 |
| | Scheme 4 Total synthesis of andirolactone 17. | |
Conclusions
In summary, we have developed a novel, highly regio- and/or diastereoselective tertiary allylic acetylation of geminally disubstituted olefins using Hg(OAc)2. This reaction method was applied for the total synthesis of andirolactone with a very good overall yield. Although the catalyst used in the above mentioned transformation is not desirable due to its toxicity, the emphasis of this manuscript is on the novel transformation to regioselective forms of tert-acetates, with the chance to modify/change to a better catalyst in future investigations. Presently, there are ongoing efforts in our lab to replace toxic Hg(OAc)2 with a better catalyst for this transformation.
Acknowledgements
B. D. and R. B. thank CSIR, New Delhi. Financial support from DST, New Delhi (project no. SB/S1/OC-01/2014) is gratefully acknowledged.
Notes and references
-
(a) D. H. Dethe and G. Murhade, Org. Lett., 2013, 15, 429 CrossRef CAS PubMed;
(b) D. H. Dethe and G. M. Murhade, Chem. Commun., 2013, 49, 8051 RSC.
- W. Treibs, Justus Liebigs Ann. Chem., 1953, 581, 59 CrossRef CAS.
-
(a) R. L. Danheiser, R. G. Brisbois, J. J. Kowalczyk and R. F. Miller, J. Am. Chem. Soc., 1990, 112, 3093 CrossRef CAS;
(b) N. Agenet, O. Buisine, F. Slowinski, V. Gandon, C. Aubert and M. Malacria, Org. React., 2007, 68, 1 Search PubMed.
- As mercury(II)acetate is highly toxic, and it releases toxic mercury in the reaction medium, proper care must be taken while handling it and performing the reaction.
-
(a) Z. Rappoport, P. D. Sleezer, S. Winstein and W. G. Young, Tetrahedron Lett., 1965, 42, 3719 CrossRef;
(b) Z. Rappoport, L. K. Dyall, S. Winstein and W. G. Young, Tetrahedron Lett., 1970, 3483 CrossRef CAS;
(c) Z. Rappoport, S. Winstein and W. G. Young, J. Am. Chem. Soc., 1972, 94, 2320 CrossRef CAS;
(d) T. R. Hoye and M. J. Rother, J. Org. Chem., 1979, 44, 458 CrossRef CAS.
-
(a) J. E. McMurry and P. Kocotovsky, Tetrahedron Lett., 1984, 25, 4187 CrossRef CAS;
(b) S. Bystroem, E. M. Larsson and B. Aakermark, J. Org. Chem., 1990, 55, 5674 CrossRef CAS;
(c) M. S. Chen and M. Christina White, J. Am. Chem. Soc., 2004, 126, 1346 CrossRef CAS PubMed.
- M. S. Kharasch, G. Sosnovsky and N. C. Yang, J. Am. Chem. Soc., 1959, 81, 5819 CrossRef CAS.
- H. P. Jensen and K. B. Sharpless, J. Org. Chem., 1975, 40, 264 CrossRef CAS.
- H. Avcibasi, H. Anil and M. Toprak, Phytochemistry, 1987, 26, 2852 CrossRef CAS.
-
(a) A. Srikrishna and G. V. R. Sharma, Tetrahedron Lett., 1988, 29, 6487 CrossRef CAS;
(b) N. Krause, Liebigs Ann. Chem., 1990, 603 CrossRef CAS;
(c) R. C. Larock, D. E. Stinn and M. Y. Kuo, Tetrahedron Lett., 1990, 31, 170 CrossRef;
(d) A. Orduna, G. Zepeda and J. Tamariz, Synthesis, 1993, 375 CrossRef CAS;
(e) P. Quayle and E. L. Ward, Tetrahedron Lett., 1994, 35, 8883 CrossRef CAS;
(f) M. Planas, C. Segura, M. Ventura and R. M. Ortuno, Synth. Commun., 1994, 24, 651 CrossRef CAS;
(g) Y. Li, T. Zhang and Y.-L. Li, Tetrahedron Lett., 2007, 48, 1503 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00310a |
|
| This journal is © the Partner Organisations 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.