
Direct amidation of the phenylalanine moiety in short peptides via Pd-catalyzed C–H activation/C–N formation†
Mingyu
Yang
a,
Xingyu
Jiang
a and
Zhang-Jie
Shi
*ab
aBeijing National Laboratory of Molecular Sciences (BNLMS) and Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry and Green Chemistry Center, Peking University, Beijing, 100871, China. E-mail: zshi@pku.edu.cn; Web: http://www.shigroup.cn/ Fax: (+86)-10-6276-0890
bState Key Laboratory of Organometallic Chemistry, Chinese Academy of Sciences, Shanghai, 200032, China
First published on 28th November 2014
Abstract
Selective modifications of life's basic components, such as polypeptides, are of great importance in both chemistry and biology. Herein, we have developed an unprecedented intramolecular amidation of the phenylalanine moiety in dipeptides via Pd-catalyzed sp2 C–H activation and C–N formation.
Selective modifications of life's basic components via chemical protocols lay the foundation for building designed organisms from the ground up and achieving conventional chemical reactions in vivo, which are two of the ultimate aims of synthetic chemistry in life sciences. Since proteins are one of the most vital and abundant biomacromolecules for life, their polypeptide structural feature continues to be an active research object for synthetic chemists. However, tough challenges are encountered if planning to use peptides as substrates for purely chemical transformations without the help of enzymes, due to solubility issues and uncontrollable selectivity. For example, realizing the chemo- and regioselectivity of designed chemical transformations on complicated peptides containing manifold functional groups as well as various reactive sites is a formidable challenge.1 As one of the tools available to functionalize organic molecules, transition metal catalysis may not be a good choice, since transition metal catalysts are readily deactivated by the detrimental chelation of substrate heteroatoms if spatially matched with the peptide as a research subject.2 Secondary, tertiary and even higher ordered structures also hamper the selective functionalization of peptides. Last but not least, the solubility and thermostability of peptides under traditional chemical conditions also remain significant concerns.
In the past several years, the in situ functionalization of peptides, as well as some special proteins, has been studied, and some exciting chemistry has been developed by prominent researchers despite facing exceeding difficulties. For example, suitable bioorthogonal ligation reactions, which could afford the covalent fusion of biomolecules bearing specific functional groups, provide excellent labelling tools.3,4 Besides the modification of active functional groups in peptides, the inactive substituents in the natural residues of peptides have hardly been approached. As transition metal catalyzed reaction is one of the most powerful methods to diversify molecules, some methods employing transition metal catalysts have been adapted to modify short peptides.5 Therefore, other unexplored adaptations of conventional chemical reactions catalyzed by transition metals for peptides as well as proteins are regarded as promising research targets of high value. Herein, we present the first intramolecular amidative annulation to convert the phenylalanine moiety of short peptides into a cyclic unnatural amino residue via Pd catalysis.
C–H functionalization has been a hotspot of research for years because of its attractive atom- and step-economy, synthetic efficiency and novelty in fundamental chemical research. Various chemical bonds, such as C–C,6 C–N,7 C–O,8 C–B9 and C–Si10 can be constructed readily and directly starting from C–H bonds. Undoubtedly, the implementation of C–H functionalization strategies to life's basic substances possesses great importance, for it will greatly expand the scope of application of C–H functionalization reactions, and enrich the toolkit of biologists for peptide modification. Due to the innate diversity and complexity of peptides, a large library of modified peptides with potential biochemical applications could be produced in a quick and convenient fashion through direct and selective C–H functionalization.
Some pioneering methods for the site-specific functionalization of short peptides have been reported in the past few decades. However, most of them only focused on the reaction of the most active α-position of the glycine moiety1,11 and the 2-position of the tryptophan ring.12 Indeed, a beautiful example of the Pd-catalyzed direct C–H functionalization of the tryptophan residue of proteins has been well studied. More complementary methods enabling selective modifications on different sites of peptides via C–H activation are in demand.
As a subfield of C–H functionalization, sp2 C–H activation and intramolecular C–N bond formation sequences could furnish a variety of N-containing ring systems in a swift manner, thus exhibiting a supplementary approach to the powerful Buchwald–Hartwig amination reaction.7a,13 Enlightened by these elegant pioneering works and with an eye on the ubiquity of the phenylalanine moiety on polypeptide chains in nature, we conceived that it would be very possible and useful to perform the unprecedented intramolecular amidation of the phenylalanine moiety in small peptides via palladium catalysis. This proposed transformation would serve as a latent instrument for the chemical modification of peptides with excellent feasibility and practicability. Most importantly, a proline-like 2,3-dihydroindolyl moiety could be constructed after the envisioned amidation, which would result in a beta-turn-type twist in the linear chain and induce a complete change in the secondary structure of the peptide (Scheme 1).14 Therefore, this work could cast a light on a prospective method for the artificial and chemical adjustment of the conformation of polypeptides, if applicable.
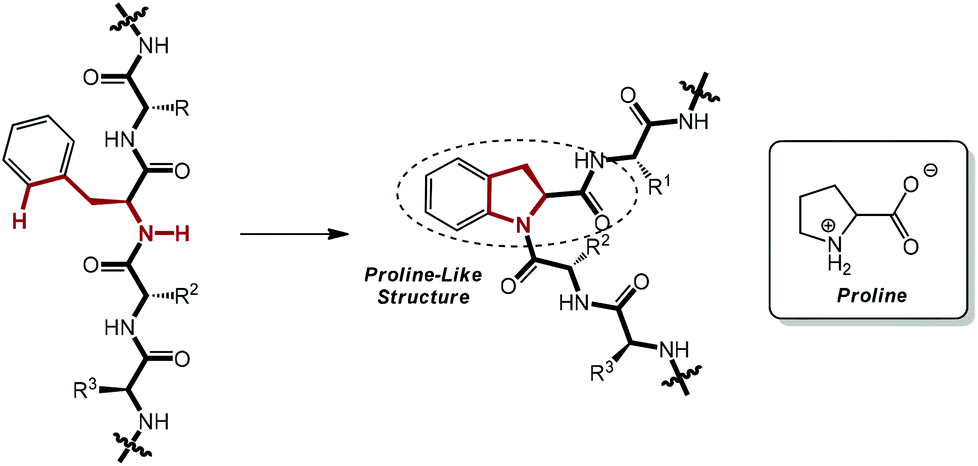 | ||
| Scheme 1 A linear peptide chain would be twisted after the envisioned selective intramolecular reaction. | ||
We started our research with the simple dipeptide 1aa containing phenylalanine for the optimization of reaction conditions. Initial screening of common oxidants used in Pd chemistry unfortunately gave less than 5% of 2aa (Table 1, entries 1–7). To our delight, with a higher loading of Ce(SO4)2, a modest yield of the desired product was obtained (entries 8–9). However, a further increase in equivalents of Ce(SO4)2 (8.0 equiv.) is not beneficial for the efficiency or yield (entry 10). Changes of ligand or equivalents of DMF failed to give better results, while the reaction was even impeded with excessive DMF (entries 11–14). Unfortunately, inconsistent results were observed among multiple experiments when 6.0 equivalents of Ce(SO4)2 and 6.0 equivalents of DMF were added (entry 9). After many attempts to stabilize the result, we were pleased to find that the addition of TsOH led to a lower but reproducible yield (entry 15). Further screening of acids revealed that MsOH was the best (entries 16–19), and reducing the amount of MsOH had little impact on the reaction, while even a slightly higher yield was obtained (entries 20–21).
|
|
||||
|---|---|---|---|---|
| Entry | Oxidant | Additive | Acid | Yielda (%) |
| a 1H NMR yield with CH2Br2 as internal standard. b Isolated yield in parentheses. c Without acid. d A is 2-nitrobenzenesulfonic acid. | ||||
| 1 | PhI(OAc)2 (2.0 eq.) | DMF (6.0 eq.) | —c | 10 |
| 2 | K2S2O8 (2.0 eq.) | DMF (6.0 eq.) | —c | 14 |
| 3 | Cu(OAc)2 (2.0 eq.) | DMF (6.0 eq.) | —c | <5 |
| 4 | TBP (2.0 eq.) | DMF (6.0 eq.) | —c | <5 |
| 5 | Oxone (2.0 eq.) | DMF (6.0 eq.) | —c | <5 |
| 6 | NFSI (2.0 eq.) | DMF (6.0 eq.) | —c | 16 |
| 7 | Ce(SO4)2 (2.0 eq.) | DMF (6.0 eq.) | —c | 37 |
| 8 | Ce(SO4)2 (4.0 eq.) | DMF (6.0 eq.) | —c | 49 |
| 9 | Ce(SO4)2 (6.0 eq.) | DMF (6.0 eq.) | —c | 56 |
| 10 | Ce(SO4)2 (8.0 eq.) | DMF (6.0 eq.) | —c | 48 |
| 11 | Ce(SO4)2 (6.0 eq.) | DMF (3.0 eq.) | —c | 55 |
| 12 | Ce(SO4)2 (6.0 eq.) | DMF (12.0 eq.) | —c | 29 |
| 13 | Ce(SO4)2 (6.0 eq.) | NMP (6.0 eq.) | —c | 27 |
| 14 | Ce(SO4)2 (6.0 eq.) | DMAc (6.0 eq.) | —c | 42 |
| 15 | Ce(SO4)2 (6.0 eq.) | DMF (6.0 eq.) | TsOH (1.0 eq.) | 47 |
| 16 | Ce(SO4)2 (6.0 eq.) | DMF (6.0 eq.) | A (1.0 eq.) | 40 |
| 17 | Ce(SO4)2 (6.0 eq.) | DMF (6.0 eq.) | TFA (1.0 eq.) | 45 |
| 18 | Ce(SO4)2 (6.0 eq.) | DMF (6.0 eq.) | AcOH (1.0 eq.) | 23 |
| 19 | Ce(SO4)2 (6.0 eq.) | DMF (6.0 eq.) | MsOH (1.0 eq.) | 57 |
| 20 | Ce(SO4)2 (6.0 eq.) | DMF (6.0 eq.) | MsOH (0.5 eq.) | 60 |
| 21 | Ce(SO4)2 (6.0 eq.) | DMF (6.0 eq.) | MsOH (0.2 eq.) | 60 (53) |
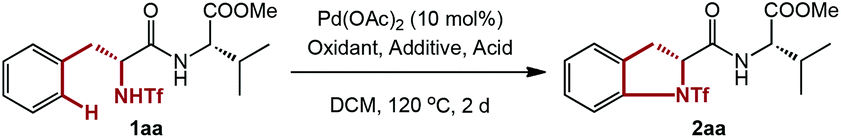
Having the optimized reaction conditions in hand, several substrates with common N-protecting groups other than Tf were subjected to the standard conditions (Table 2). However, the desired products were detected in less than 5% yields when using Ts, TFA, Boc or Ac protecting groups, manifesting the unique activity of triflamide.13c–e,15

To test the effect of the relative stereochemistry in the dipeptide substrate, all 4 diastereoisomers of dipeptide 1aa were synthesized and submitted to the standard conditions (Table 3). In fact, similar results were obtained, showing little difference in the reactivity of these 4 isomers with different diastereochemistry (2aa–2ad). This discovery substantiated the negligible effect of the configuration of the substrate on the reaction, and rationalized the utilization of several substrates with configurations different to that of 1aa for further study.
| a Yield based on isolated yield divided by percentage of conversion. |
|---|
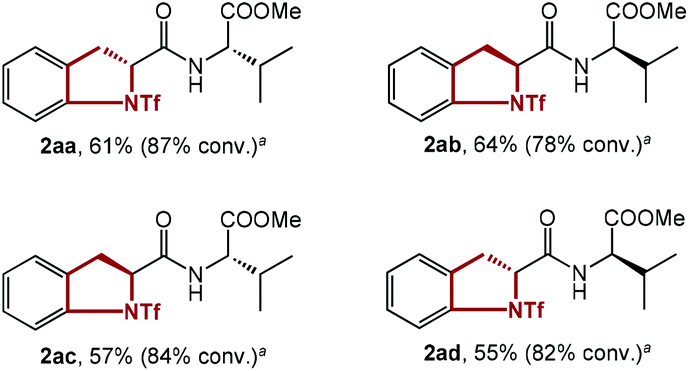
|
To determine the feasibility of this transformation, various dipeptides containing phenylalanine and another naturally occurring amino acid were studied (Table 4). To our interest, the steric effect of the R group had a notable impact on the efficiency of the amidation. For example, no product was detected when the chain of the amino acid residue at the C-terminus was glycine (2b). Along with the extension of the amino acid side chain from a methyl to an isobutyl group, all the amidations occurred with comparable efficacy (2c–2e). A branched structure on the beta position of the C-terminus further promoted the yield significantly (2aa, 2g). A Phe–Phe dipeptide was also tested, and the expected annulation took place exclusively on the side of the triflamide (2f). It is worth noting that although much lower yields were afforded, the catalyst maintained its activity in the presence of two ester groups at the C-terminus, where excessive and deleterious coordination to the palladium center was possible (2h–2i).
| a Conditions: 1 (0.1 mmol), Pd(OAc)2 (2.2 mg, 0.1 equiv.) and Ce(SO4)2 (199.0 mg, 6.0 equiv.) were placed in a vial under air. DCM (0.8 mL), DMF (47 μL, 6.0 equiv.) and MsOH (0.2 mL of a 0.1 M solution in DCM) were added sequentially, the mixture was maintained at rt for 5 min and then stirred for 2 d at 120 °C. b Yield based on isolated yield divided by percentage of conversion. c The configuration at the carbon where the methyl and ethyl group are attached is S. |
|---|

|
Many dipeptides with unnatural amino acids at the C-terminus, which might serve as the key reactive part in peptide modification,16 were also well tolerated in our system, showing that continuous and multiple modifications on peptides are possible. Consistent with the results shown above, a dipeptide with a large t-butyl group as a side chain afforded a good yield (2j). To further prove this trend, 2-methylalanine was used as the C-terminal amino acid, which may promote the efficiency by the Thorpe-Ingold effect. Although a larger steric hindrance was induced through the installation of two methyl groups, an unexpected decrease in yield was discovered (2k), which might be attributed to the increased steric strain and improper configuration of the substrate, causing the inner stability of the intermediate to deteriorate. Substrates with O-esterificated 2-amino alcohols at the C-terminus, which could be obtained readily by a simple reduction from amino acids, also gave good yields (2m–2n). Thus, a variety of potential substrates of the type 2m are accessible in a straightforward manner by taking advantage of plentiful amino acids.
Comparing the different substrates with different C-terminal moieties (2k, 2l, 2m and 2o), the results clearly illustrated the necessity of the presence of an ester group at the C-terminus for satisfying reactivity, which may arise from the possible coordination of the carbonyl group of the ester to the amino alcohol instead of the ether group of the dipeptide, stabilizing the key intermediate. This conclusion is consistent with the similar “relay effect” of appropriately positioned heteroatoms in transition metal-catalyzed transformations which has been reported previously.17 Finally, experiments targeting the electronic effect of the phenyl ring on this transformation were conducted. Surprisingly, only electron-neutral substrates underwent the amidation smoothly with high conversion and yield, while both electron-donating groups and electron-withdrawing groups dramatically diminished the efficacy, and an electron-rich substrate was over-oxidized to an indole structure (2p–2q).
Another goal when modifying peptides is to retain the optical purity during transformations. Traditional methods for functionalizing short peptides usually require the use of excess strong base to form an enolate intermediate, which leads to racemization at the stereogenic centers of peptides. In our developed method, the reaction conditions are relatively mild, with the absence of either strong base or acid. Indeed, to our satisfaction, under the developed conditions, diverse optically pure N-triflated short peptides (except 2p) were tested, and the corresponding single diastereoisomers were isolated without any racemization, demonstrating the retention of configuration for modified short peptides.
Conclusions
In summary, we have developed the first direct amidation of dipeptides through direct aryl C–H transformation of the phenylalanine residue via Pd-catalysis with good efficacy. This cyclization exhibited flexibility with different amino acids at the C-terminus. In the displacement of amino acid esters by aminoalcohol acetates, good efficacy was obtained, thus extending the substrate scope. Under such mild conditions, no racemization of either of the amino acids of the dipeptide was observed. Further studies to improve the efficacy and explore such chemistry for long peptides and even proteins are under way.Acknowledgements
The authors gratefully acknowledge support for this work the “973” project from the MOST of China (2015CB856600) and NSFC (grant nos. 21431008 and 21332001).Notes and references
- L. Zhao, O. Baslé and C.-J. Li, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4106 CrossRef CAS PubMed.
- M. A. Larsen and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 4287 CrossRef CAS PubMed.
- (a) J. A. Prescher, D. H. Dube and C. R. Bertozzi, Nature, 2004, 430, 873 CrossRef CAS PubMed; (b) J. A. Prescher and C. R. Bertozzi, Nat. Chem. Biol., 2005, 1, 13 CrossRef CAS PubMed; (c) E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974 CrossRef CAS PubMed; (d) E. M. Sletten and C. R. Bertozzi, Acc. Chem. Res., 2011, 44, 666 CrossRef CAS PubMed.
- R. K. V. Lim and Q. Lin, Chem. Commun., 2010, 46, 1589 RSC.
- T. Plass and C. Schultz, Springer Ser. Fluoresc., 2011, 113, 225 Search PubMed.
- For recent reviews, see: (a) J. Bouffard and K. Itami, Top. Curr. Chem., 2010, 292, 231 CrossRef CAS; (b) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (c) L. L. Bras and J. Muzart, Chem. Rev., 2011, 111, 1170 CrossRef PubMed; (d) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (e) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (f) D.-G. Yu, B.-J. Li and Z.-J. Shi, Tetrahedron, 2012, 68, 5130 CrossRef CAS PubMed.
- (a) W. C. P. Tsang, N. Zheng and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 14560 CrossRef CAS PubMed; (b) X. Chen, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790 CrossRef CAS PubMed; (c) H.-Y. Thu, W.-Y. Yu and C.-M. Che, J. Am. Chem. Soc., 2006, 128, 9048 CrossRef CAS PubMed; (d) T. Tan and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 3676 CrossRef PubMed; (e) C. Grohmann, H. Wang and F. Glorius, Org. Lett., 2012, 14, 656 CrossRef CAS PubMed; (f) G. He, C. Lu, Y. Zhao, W. A. Nack and G. Chen, Org. Lett., 2012, 14, 2944 CrossRef CAS PubMed.
- (a) L. V. Desai, K. J. Stowers and M. S. Sanford, J. Am. Chem. Soc., 2008, 130, 13285 CrossRef CAS PubMed; (b) Y.-H. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 14654 CrossRef CAS PubMed; (c) C.-L. Sun, J. Liu, Y. Wang, X. Zhou, B.-J. Li and Z.-J. Shi, Synlett, 2011, 883 CAS.
- (a) J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864 CrossRef CAS PubMed; (b) L.-S. Zhang, G.-H. Chen, X. Wang, Q.-Y. Guo, X.-S. Zhang, F. Pan, K. Chen and Z.-J. Shi, Angew. Chem., Int. Ed., 2014, 53, 3899 CrossRef CAS PubMed.
- (a) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed; (b) C. Cheng and J. F. Hartwig, Science, 2014, 343, 853 CrossRef CAS PubMed.
- (a) C. J. Easton, C. A. Hutton, G. Rositano and E. W. Tan, J. Org. Chem., 1991, 56, 5614 CrossRef CAS; (b) M. J. O'Donnell, C.-Y. Zhou and W. L. Scott, J. Am. Chem. Soc., 1996, 118, 6070 CrossRef; (c) M. J. O'Donnell, M. D. Drew, R. S. Pottorf and W. L. Scott, J. Comb. Chem., 2000, 2, 172 CrossRef; (d) K. Maruoka and T. Ooi, Chem. Rev., 2003, 103, 3013 CrossRef CAS PubMed; (e) T. Hashimoto and K. Maruoka, Chem. Rev., 2007, 107, 5656 CrossRef CAS PubMed.
- (a) J. Ruiz-Rodríguez, F. Albericio and R. Lavilla, Chem. – Eur. J., 2010, 16, 1124 CrossRef PubMed; (b) H.-J. Dong, C. Limberakis, S. Liras, D. Price and K. James, Chem. Commun., 2012, 48, 11644 RSC.
- (a) J. M. Janey, Name Reactions of Functional Group Transformations, ed. J. J. Li and E. J. Corey, Wiley, New York, 2007, pp. 564–609 Search PubMed; (b) K. Inamoto, T. Saito, M. Katsuno, T. Sakamoto and K. Hiroya, Org. Lett., 2007, 9, 2931 CrossRef CAS PubMed; (c) T.-S. Mei, X.-S. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 10806 CrossRef CAS PubMed; (d) J.-J. Li, T.-S. Mei and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 6452 CrossRef CAS PubMed; (e) T.-S. Mei, D.-S. Leow, H. Xiao, B. N. Laforteza and J.-Q. Yu, Org. Lett., 2013, 15, 3058 CrossRef CAS PubMed. For related copper-catalyzed C–H aminations, see: (f) G. Brasche and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 1932 CrossRef CAS PubMed.
- S. K. Brahmachari, R. S. Rapaka, R. S. Bhatnagar and V. S. Ananthanarayanan, Biopolymers, 1982, 21, 1107 CrossRef CAS.
- (a) L. Chu, X.-C. Wang, C. E. Moore, A. L. Rheingold and J.-Q. Yu, J. Am. Chem. Soc., 2013, 135, 16344 CrossRef CAS PubMed; (b) K. S. L. Chan, M. Wasa, L. Chu, B. N. Laforteza, M. Miura and J.-Q. Yu, Nat. Chem., 2014, 6, 146 CrossRef CAS PubMed.
- K.-H. Wang, A. Sachdeva, D. J. Cox, N. W. Wilf, K. Lang, S. Wallace, R. A. Mehl and J. W. Chin, Nat. Chem., 2014, 6, 393 CrossRef CAS PubMed.
- (a) A. Fürstner and K. Langemann, Synthesis, 1997, 792 CrossRef PubMed; (b) Y. A. Lin, J. M. Chalker, N. Floyd, G. J. L. Bernardes and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 9642 CrossRef CAS PubMed; (c) M. M. Coulter, P. K. Dornan and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 6932 CrossRef CAS PubMed; (d) C. González-Rodríguez and M. C. Willis, Pure Appl. Chem., 2011, 83, 577 CrossRef; (e) A. P. Thottumkara, T. Kurokawa and J. Du Bois, Chem. Sci., 2013, 4, 2686 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4qo00282b. |
| This journal is © the Partner Organisations 2015 |