
Recent applications of chiral N-tert-butanesulfinyl imines, chiral diene ligands and chiral sulfur–olefin ligands in asymmetric synthesis
Han-Qing
Dong†
a,
Ming-Hua
Xu
*b,
Chen-Guo
Feng
a,
Xing-Wen
Sun
c and
Guo-Qiang
Lin
*a
aCAS Key Laboratory of Synthetic Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: lingq@sioc.ac.cn
bState Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China. E-mail: xumh@simm.ac.cn
cDepartment of Chemistry, Fudan University, 220 Handan Road, Shanghai 200433, China
First published on 18th November 2014
Abstract
This review highlights the recent applications of chiral N-tert-butanesulfinyl imines and chiral diene ligands, with bicyclo[3.3.0]octadiene and dicyclopentadiene skeletons, in asymmetric chemical transformations. The chiral sulfinamide–olefin products from allylation of N-tert-butanesulfinyl imines can be used as hybrid ligands for transition metal-catalyzed asymmetric reactions. The efforts in the further exploration of chiral sulfur–olefin ligands are also discussed.
 Han-Qing Dong | Han-Qing Dong obtained his Ph.D. in organic chemistry from the Shanghai Institute of Organic Chemistry (SIOC), Chinese Academy of Sciences (CAS) in 1997. He was awarded the Alexander von Humboldt (AvH) fellowship and performed research in Prof. Volker Jäger's laboratory at the University of Stuttgart in Germany (1998–1999). Subsequently, he moved to the USA and conducted post-doctoral research in Prof. Andrew S. Kende's group at the University of Rochester (1999–2001). Dr Dong has extensive experience in synthetic organic chemistry and drug discovery. He has worked on multiple oncology targets at OSI Pharmaceuticals/Astellas Pharma (2002–2013), and is currently a lead chemist at Arvinas Inc. |
 Ming-Hua Xu | Ming-Hua Xu was born in Zhejiang, China. He obtained his Ph.D. from SIOC in 1999. Following postdoctoral research at the University of Virginia and Georgetown University Medical Center, he joined the faculty at SIOC as an associate professor in 2003. He has been a professor at the Shanghai Institute of Materia Medica, Chinese Academy of Sciences since 2005. His research interests mainly involve asymmetric synthesis and drug discovery. |
 Chen-Guo Feng | Chen-Guo Feng received his MSc (2001) in chemistry from Xiamen University under the supervision of Prof. Pei-Qiang Huang. After he obtained his Ph.D. (2009) from SIOC under the supervision of Prof. Guo-Qiang Lin, he spent two years at the Scripps Research Institute (La Jolla, CA) with Prof. Jin-Quan Yu as a postdoctoral fellow. He is currently an associate professor in SIOC and his research interests include asymmetric catalysis and medicinal chemistry. |
 Xing-Wen Sun | Xing-Wen Sun was born in Henan province, China. He received his undergraduate education at the Zhengzhou University and graduated in 2002. In the fall of that year, Xing-Wen enrolled in graduate studies at SIOC and joined the research group of Prof. Guo-Qiang Lin. He earned his Ph.D. in organic chemistry in 2007. In July 2007, he began his independent career at Fudan University, where his research program focuses on the total synthesis of natural products and the development of new methods of organic synthesis. |
 Guo-Qiang Lin | Guo-Qiang Lin pursued his graduate studies (1964–1968) in SIOC with Prof. Wei-Shan Zhou. He has been a professor in the same institute since 1990. His current research interests include the synthesis of natural products and biologically active compounds, asymmetric catalysis and biotransformation. |
1. Introduction
Asymmetric synthesis with both high level of selectivities and yields remains a challenge in spite of significant achievements in the last few decades.1 Chiral N-tert-butanesulfinyl imines have been intensively investigated to generate enantioenriched 1,2-diamines and β-amino alcohols through SmI2-induced homo-coupling and cross-coupling with nitrones and aldehydes. With our discovery of these useful transformations, we demonstrated their synthetic applicability in the total syntheses of (3R,4S)-statine, D-erythro-sphinganine, (+)-febrifugine, (+)-CP-99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 994 and (+)-L-733060. Additionally, we studied the Zn/In-mediated allylation of N-tert-butanesulfinyl imines to provide a variety of chiral homoallylic amines.2 In the meantime, we discovered a series of bicyclo[3.3.0]octadiene and dicyclopentadiene-based ligands, and successfully employed them in rhodium-catalyzed asymmetric addition to imines, α,β-unsaturated ketones/esters, nitroalkenes and α,β-unsaturated γ-lactams.3 In this review, we highlight our recent advancement in the allylation of chiral N-tert-butanesulfinyl imines, as well as novel asymmetric reactions using our chiral diene ligands. In addition, by combining N-tert-butanesulfinamide and olefin elements into one ligand molecule, an unprecedented type of sulfur–olefin hybrid ligand has been designed. Gratifyingly, the use of chiral sulfinamide-containing allylation products as ligands in rhodium-catalyzed asymmetric reactions proved to be successful (Fig. 1). Furthermore, a variety of sulfur–olefin ligands were prepared and evaluated, and excellent enantioselectivities were achieved in a series of asymmetric arylation reactions of C
994 and (+)-L-733060. Additionally, we studied the Zn/In-mediated allylation of N-tert-butanesulfinyl imines to provide a variety of chiral homoallylic amines.2 In the meantime, we discovered a series of bicyclo[3.3.0]octadiene and dicyclopentadiene-based ligands, and successfully employed them in rhodium-catalyzed asymmetric addition to imines, α,β-unsaturated ketones/esters, nitroalkenes and α,β-unsaturated γ-lactams.3 In this review, we highlight our recent advancement in the allylation of chiral N-tert-butanesulfinyl imines, as well as novel asymmetric reactions using our chiral diene ligands. In addition, by combining N-tert-butanesulfinamide and olefin elements into one ligand molecule, an unprecedented type of sulfur–olefin hybrid ligand has been designed. Gratifyingly, the use of chiral sulfinamide-containing allylation products as ligands in rhodium-catalyzed asymmetric reactions proved to be successful (Fig. 1). Furthermore, a variety of sulfur–olefin ligands were prepared and evaluated, and excellent enantioselectivities were achieved in a series of asymmetric arylation reactions of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CO and CN bonds.4
C, CO and CN bonds.4
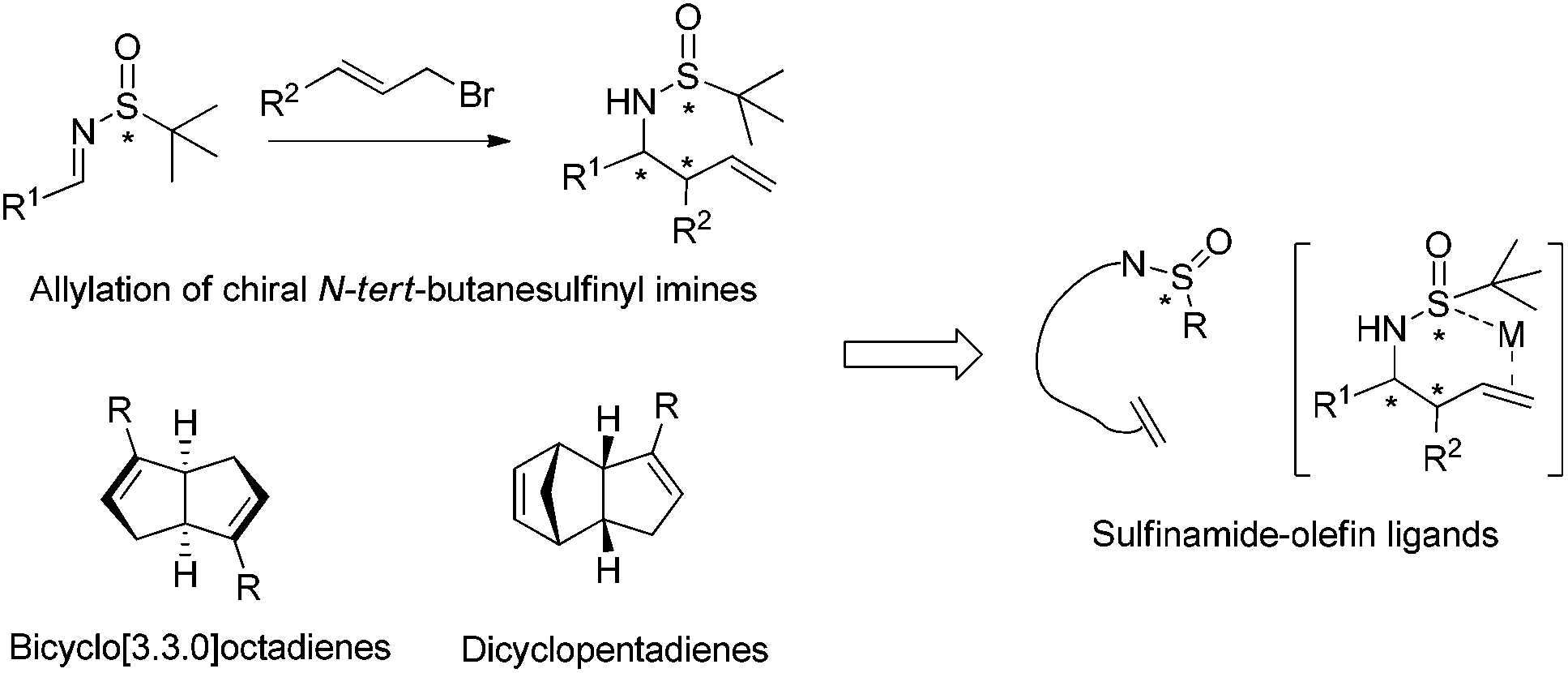 | ||
| Fig. 1 Our footprints to chiral sulfur–olefin ligands. | ||
2. Synthetic applications of chiral N-tert-butanesulfinyl imines toward α-chiral amines
Chiral N-tert-butanesulfinyl imine 1, whether in the R or S form, can be obtained from the corresponding enantiopure R or S N-tert-butanesulfinamide 2 (Fig. 2). Pioneered by Ellman and co-workers,5 both enantiomers of imine 1 were applied in the preparation of a wide range of optically active amines. The investigations in this field were rooted in the SmI2-mediated asymmetric synthesis of α,γ-substituted γ-butyrolactones.6 During the last few years, we found that imine 1 could be subjected to SmI2-mediated pinacol-type coupling reactions, Lewis acid promoted aza-Friedel–Crafts reactions, and Zn/In-mediated allylation reactions, producing a diverse range of new chiral skeletons in organic syntheses. | ||
| Fig. 2 N-tert-Butanesulfinyl imines and their synthetic precursors N-tert-butanesulfinamides. | ||
2.1. SmI2-mediated syntheses of chiral 1,2-diamines and β-amino alcohols
Samarium(II) iodide (SmI2), also known as Kagan's reagent,7 is a mild one-electron donating agent. Upon treatment with 2 equivalents of SmI2, N-tert-butanesulfinyl imines were converted to an anion, which could undergo homo-coupling to provide optically pure C2-symmetrical 1,2-diamines.8 The preparation of C2-unsymmetrical 1,2-diamines requires cross-coupling with different imines. We found that nitrones (iminium ion equivalents) could be applied to achieve our goal. In this case, a nitrone was first reduced by SmI2 to generate an α-aza nucleophilic anion, which subsequently attacked the CN bond of the N-tert-butanesulfinyl imine (Scheme 1).9 The success of cross-coupling with nitrones encouraged us to explore cross-coupling with aldehydes. Fortunately, such SmI2-mediated cross-coupling reactions could lead to the corresponding chiral β-amino alcohols in excellent diastereoselectivities and enantioselectivities.10 The synthetic values of SmI2-mediated coupling were demonstrated by the total syntheses of biologically important compounds D-erythro-sphinganine, (3R,4S)-statine and (+)-febrifugine, as well as pharmaceutical agents (+)-CP-99994 and (+)-L-733060 (Scheme 2).11
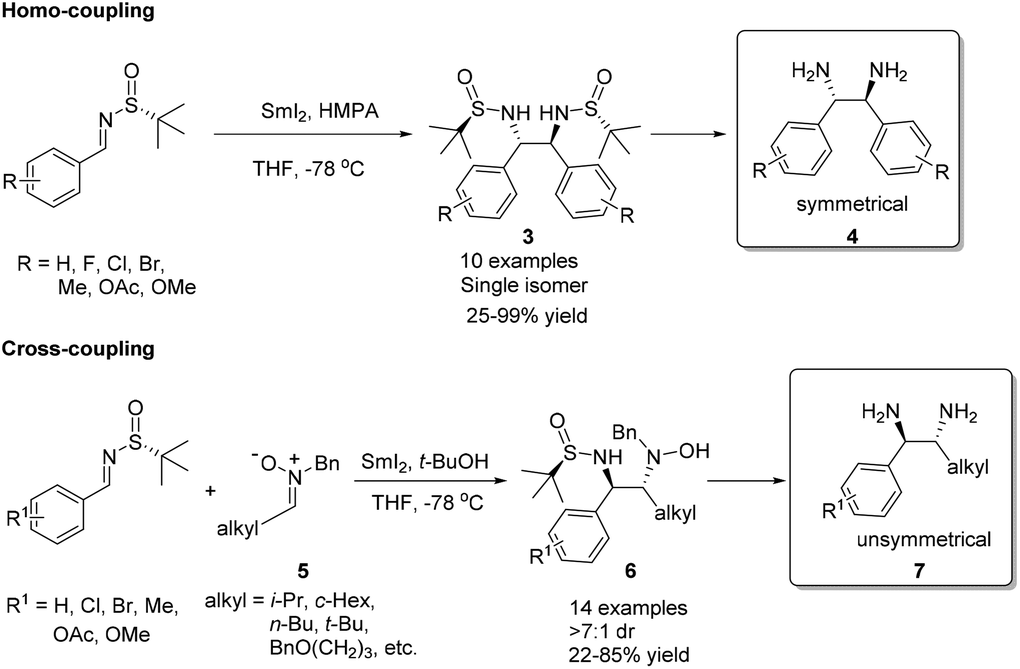 | ||
| Scheme 1 SmI2-mediated couplings to provide chiral 1,2-diamines. | ||
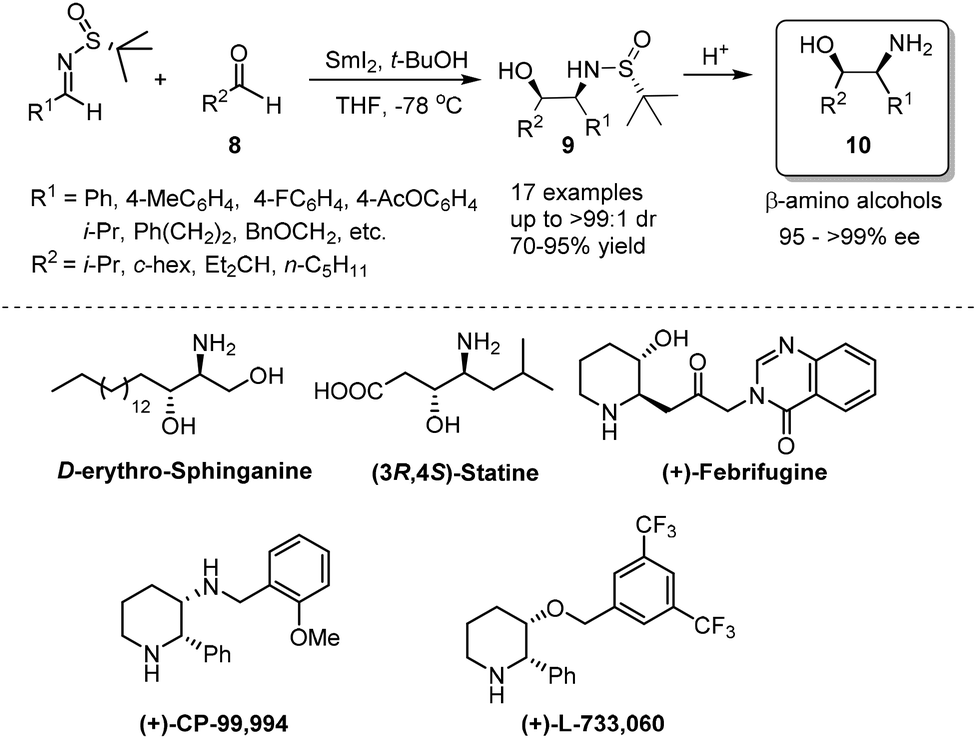 | ||
| Scheme 2 SmI2-mediated cross-coupling with aldehydes to provide chiral β-amino alcohols. | ||
2.2. Lewis acid promoted aza-Friedel–Crafts reaction of N-tert-butanesulfinyl imines
Most imines possess relatively poor electrophilicity, compared with the corresponding carbonyl compounds.12 However, N-tert-butanesulfinyl imines can provide suitable reactivity toward such nucleophilic addition in the presence of Lewis acids.Optically active non-proteinogenic amino acids are currently of great interest due to their significant biological activities, and extensive applications in organic synthesis and drug discovery.13 Asymmetric Friedel–Crafts alkylation of aromatic substrates with N-tert-butanesulfinyl glyoxylate imine 12 could be promoted by a proper transition metal-based Lewis acid catalyst, providing a convenient approach to access optically active non-proteinogenic amino acids. Under the optimal reaction conditions, N-tert-butanesulfinyl glyoxylate imine 12 could react with a wide range of indoles bearing different substituents, affording α-(3-indolyl)glycines 13 in moderate to excellent yields and with good to excellent diastereoselectivities (Scheme 3).14
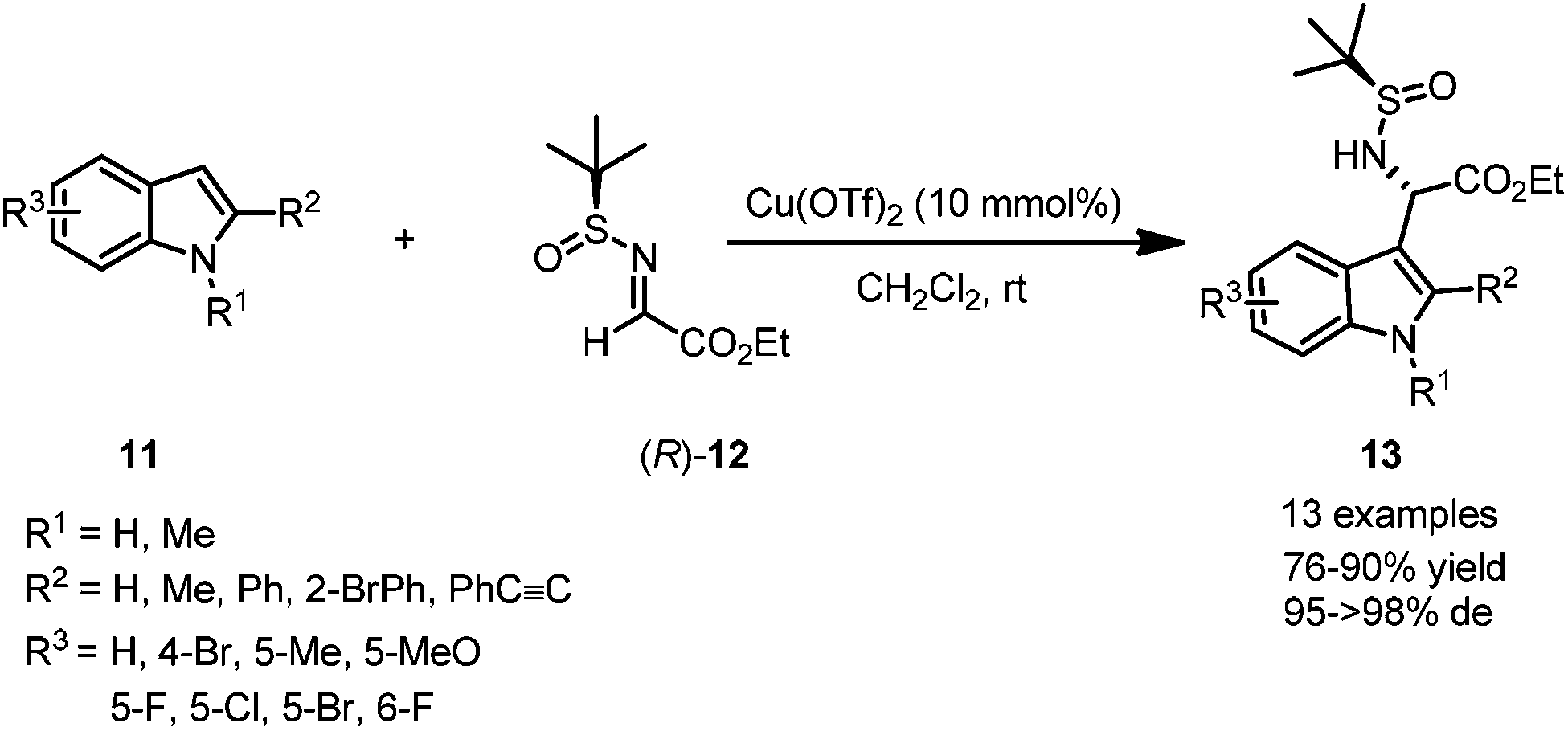 | ||
| Scheme 3 Lewis acid catalyzed asymmetric aza-Friedel–Crafts reactions of N-tert-butanesulfinyl imine to indoles. | ||
Furthermore, a variety of arenes or heteroarenes were also suitable substrates for the reaction with N-tert-butanesulfinyl glyoxylate imine 12. With In(OTf)3 as the optimal Lewis acid catalyst, the reaction could be accomplished at room temperature, providing a series of enantiomerically enriched α-arylglycines 14 in good yields and with excellent diastereoselectivities (up to 99% de) (Scheme 4). Highly stereoselective double Friedel–Crafts alkylation of 1,3-dimethoxybenzene was investigated, and the reaction proceeded smoothly, with 1 equivalent of In(OTf)3, to afford a dialkylation product in 43% yield.15
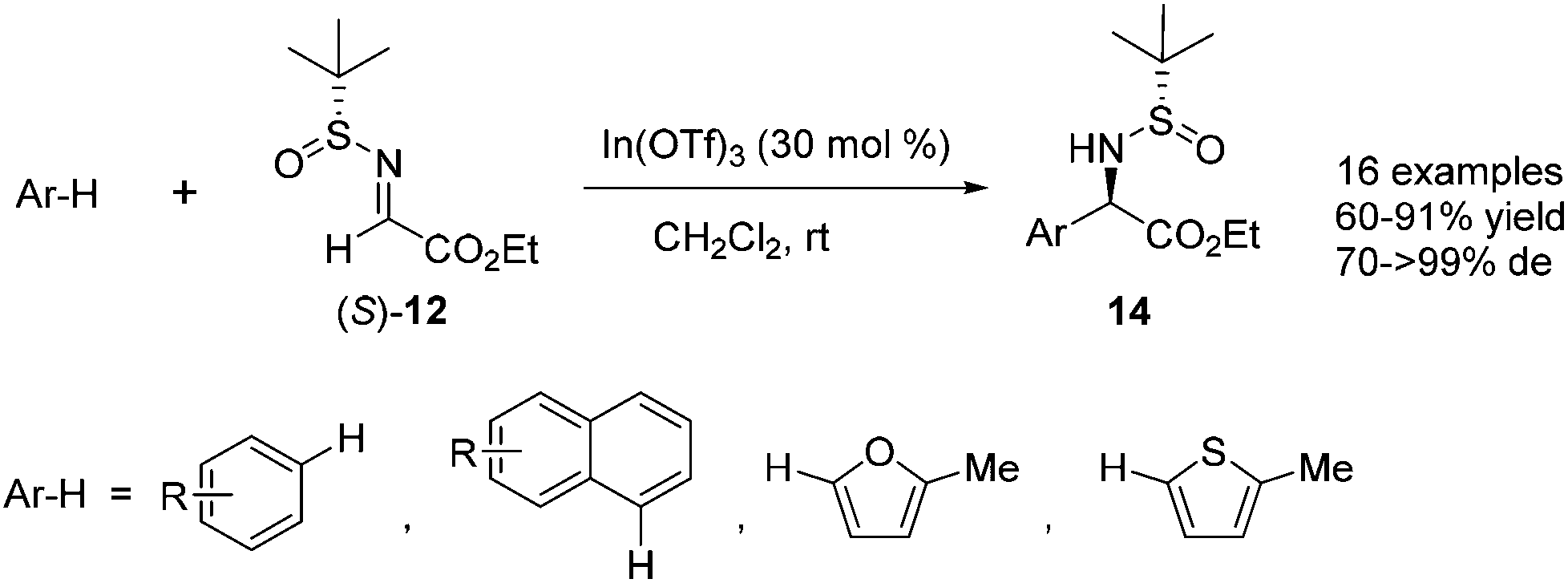 | ||
| Scheme 4 Lewis-acid-catalyzed asymmetric aza-Friedel–Crafts reactions of N-tert-butanesulfinyl imine to arenes. | ||
9-Aminofluorene and its derivatives are known as important structural motifs in biological compounds.16 A Lewis-acid-catalyzed asymmetric intramolecular aza-Friedel–Crafts reaction of N-tert-butanesulfinyl imine was designed to provide an efficient approach for the preparation of optically active 9-aminofluorenes. Bi(OTf)3 proved to be the best Lewis acid catalyst in this case, giving the desired products in good yields and with high diastereoselectivities. It is notable that both optically pure diastereomers could be separated by silica gel flash chromatography (Scheme 5).17
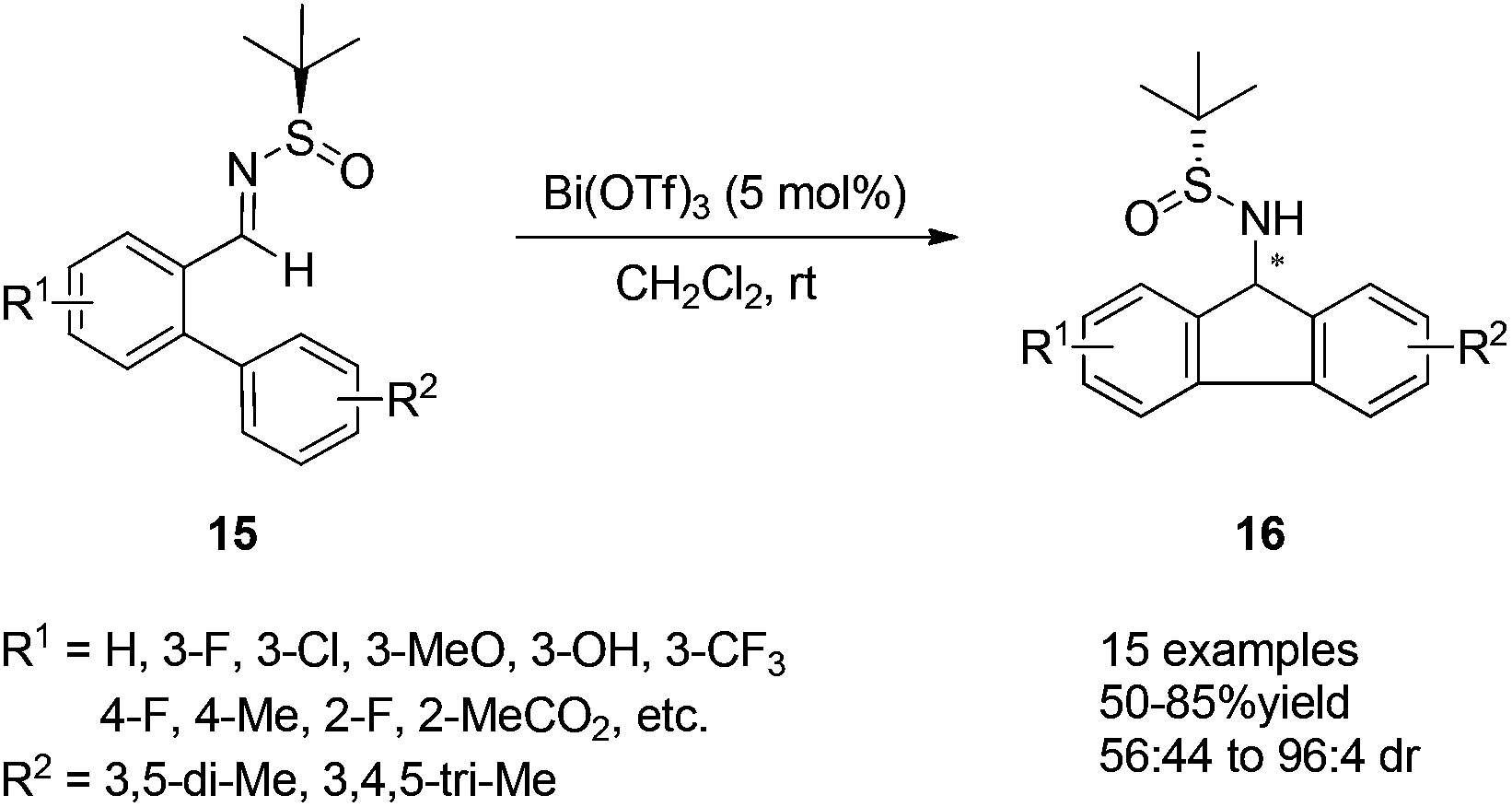 | ||
| Scheme 5 Lewis acid catalyzed asymmetric intramolecular aza-Friedel–Crafts reactions of N-tert-butanesulfinyl imines. | ||
2.3. Zn/In-mediated allylation toward chiral homoallylic amines
Chiral N-tert-butanesulfinyl imines could be subjected to the diastereoselective additions of allyl metal reagents. The Ellman group first applied allylmagnesium in addition to chiral N-tert-butanesulfinyl imines.18 Later, the Foubelo group reported the corresponding diastereoselective allylation with allylindium reagents.19 A six-membered ring transition state with metal cations was proposed to explain the high diastereoselectivities in these two reactions. Our group was also engaged in this kind of allylation, but under the Zn/In-mediated process (Scheme 6). It is worth mentioning that either enantiomeric homoallylic amine could be achieved from a common chiral N-tert-butanesulfinyl imine by slightly tuning the reaction conditions. The reaction in HMPA with a small amount of water provided one major diastereomer, while a Lewis acid additive In(OTf)3 in THF led to the preponderant formation of the other diastereomer. The two systems worked well for N-tert-butanesulfinyl imines derived from both aliphatic and aromatic aldehydes. Furthermore, the latter was successfully applied in the allylation of ketimines, giving the corresponding quaternary stereocenter-containing chiral homoallylic amines with excellent diastereoselectivities. Two new acyclic transition-states (TS-2 and TS-3), which differ from the previous six-membered chair transition-state model (TS-1), were proposed to explain the stereochemical outcomes (Scheme 6).20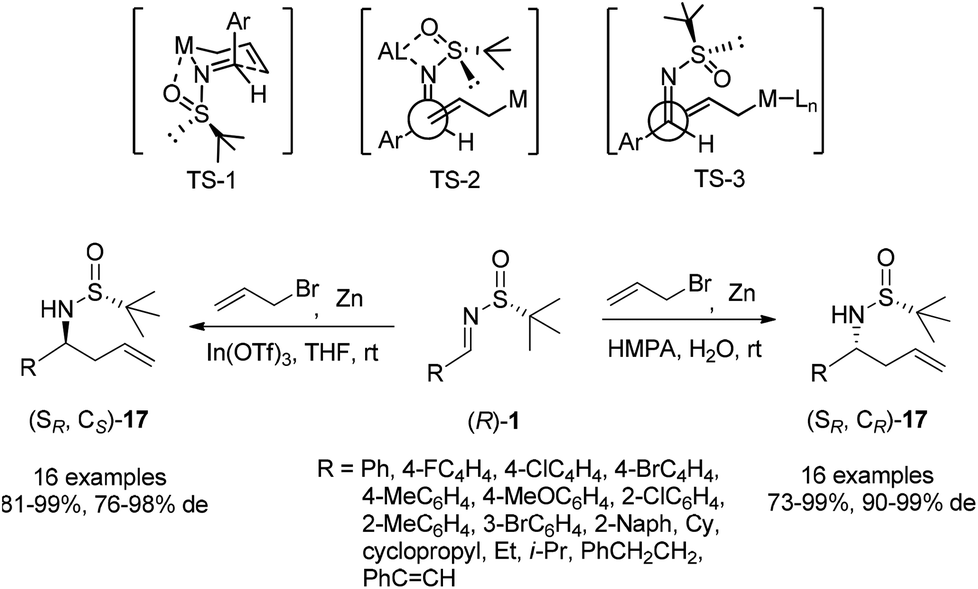 | ||
| Scheme 6 Zn-mediated allylation under two different systems. | ||
The In-mediated allylation could be conducted in aqueous media at room temperature, and saturated aqueous NaBr provided the best result. Both aryl and alkyl aldimines worked well, even in the open air, to afford the corresponding homoallylic amines 17 in high yields and with excellent diastereoselectivities (Scheme 7). The synthetic utility of this method was demonstrated by the convenient preparation of enantioenriched allylglycine, as well as the synthesis of chiral 3-allyl isoindolinones.21
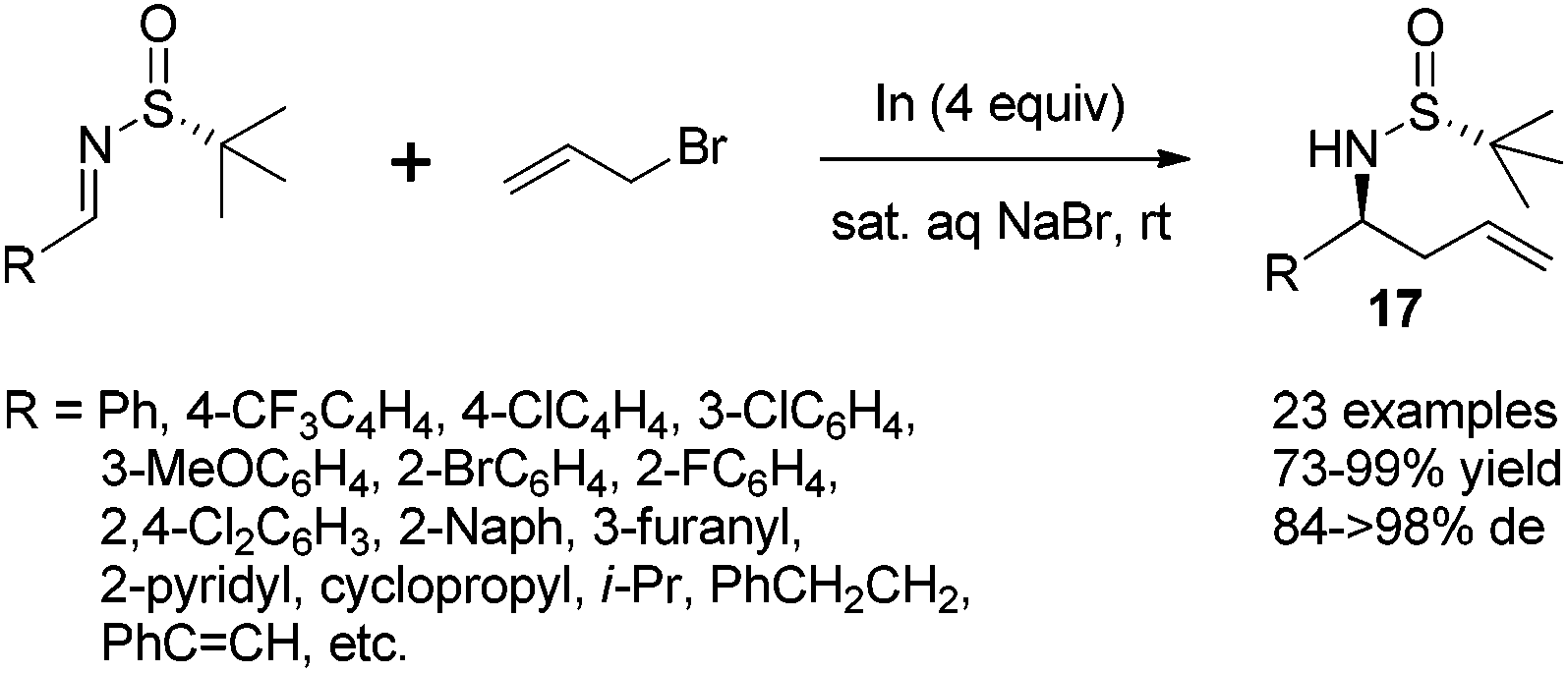 | ||
| Scheme 7 In-mediated allylation in aqueous media. | ||
The successful Zn/In-mediated allylation of N-tert-butanesulfinyl imines encouraged us to further explore the use of functionalized allylic bromide reagents, in which 3-benzoyloxyallyl bromide (3-bromopropenyl benzoate), 3-phenylallyl bromide (cinnamyl bromide), 3-methyl-2-ethoxycarbonylallylic bromide (ethyl 2-bromomethyl butenoate), as well as heterocyclic allylic bromide (3-bromomethyl-5H-furan-2-one) proved to work well, providing diverse chiral allylation products in high yields and with excellent diastereoselectivities.
The reaction of N-tert-butanesulfinyl imines with 3-benzoyloxyallyl bromide (3-bromopropenyl benzoate) 22 could generate various β-amino-α-vinyl alcohols.22 The traditional methods to prepare optically active β-amino alcohols include asymmetric aminohydroxylation of olefins, ring-opening of epoxides and aziridines, nucleophilic addition to α-hydroxyimines or α-aminocarbonyls, α-alkoxyenolate addition to imines, Mannich-type reactions of α-hydroxyketones and imines, as well as direct pinacol-type cross-coupling between carbonyls and imines.23 This new strategy offers an alternative approach as shown in Fig. 3. In this case, an α-hydroxyallyl carbanion 19 attacks an imine substrate, representing a very straightforward route to β-amino-α-vinyl alcohol derivatives 18.
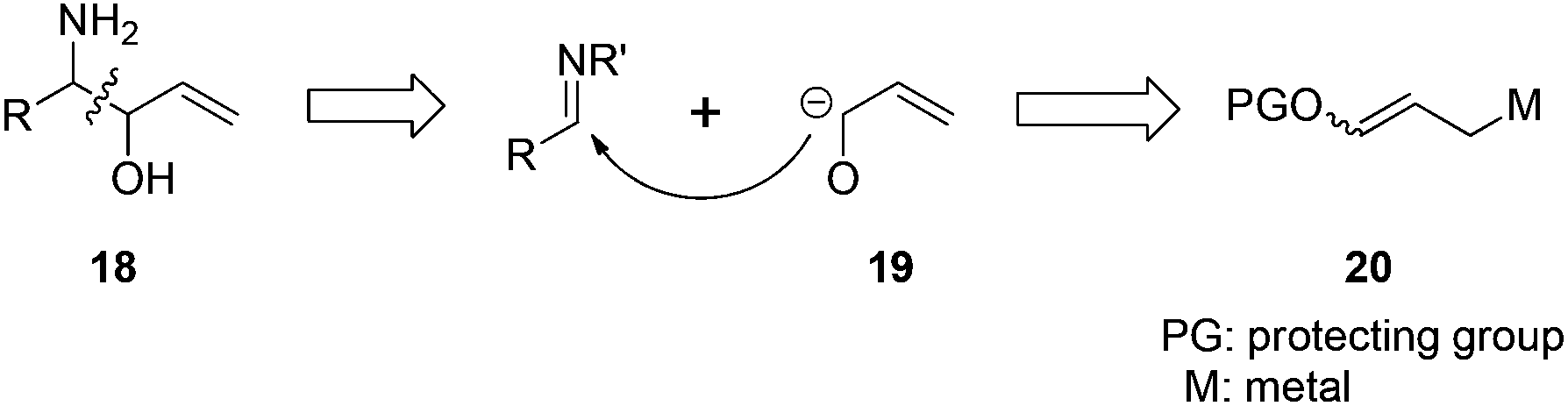 | ||
| Fig. 3 Direct α-hydroxyallylation approach. | ||
The Zn-mediated reaction of 3-benzoyloxyallyl bromide and N-tert-butanesulfinyl imines proceeded well in different solvent systems, and a reversal of stereofacial selectivity was observed again, which has been found in the simple allylic addition as shown in Scheme 8. Two different transition state models (TS-1 and TS-3) were proposed for both reaction pathways (Scheme 9). High reaction yield and stereochemical control can be achieved by using HMPA as the solvent in the presence of a small amount of water (Scheme 9). For aromatic substrates where R were different substituted phenyl or naphthyl groups, the anti/syn ratio could be achieved up to 99:1, and diastereoselectivity up to 98% de. A decreased anti/syn ratio (78:22 to 79:21) and diastereoselectivity (80–87% de) were observed when the less sterically hindered N-sulfinyl imines, derived from 3-phenylpropanal, butanal and 3-methylbutanal, were used. Interestingly, the cyclohexyl imine provided the corresponding product with the anti/syn ratio as 98:2 although the diastereoselectivity remained to be moderate (81% de). Notably, we found that the unpleasant solvent HMPA could be replaced by DMF with LiCl as an additive in this reaction.24 In addition, the use of 1,3-dibromoprop-1-ene (i.e., replacing the benzoyloxy group with bromo in compound 22) in this Zn-mediated aza-Barbier reaction was found to afford optically active trans-2-substituted vinyl aziridines.25
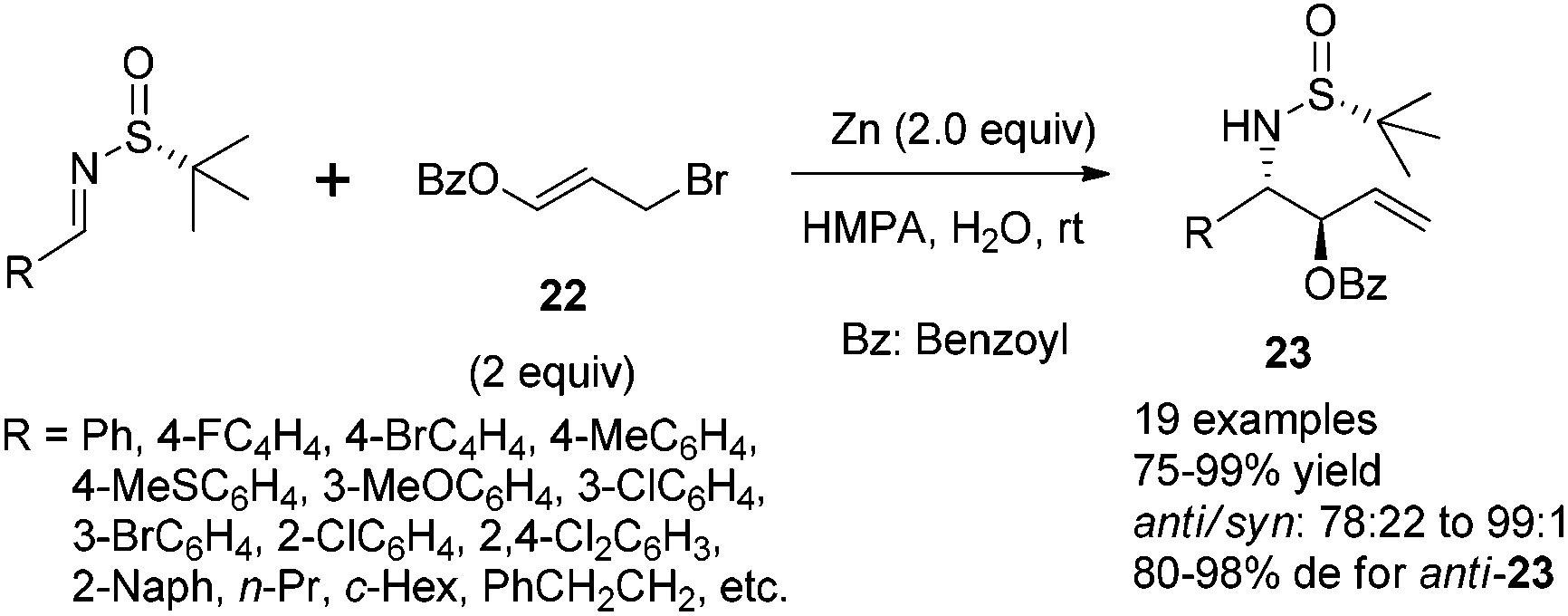 | ||
| Scheme 8 Zn-mediated benzoyloxyallylation to provide β-amino-α-vinyl alcohols. | ||
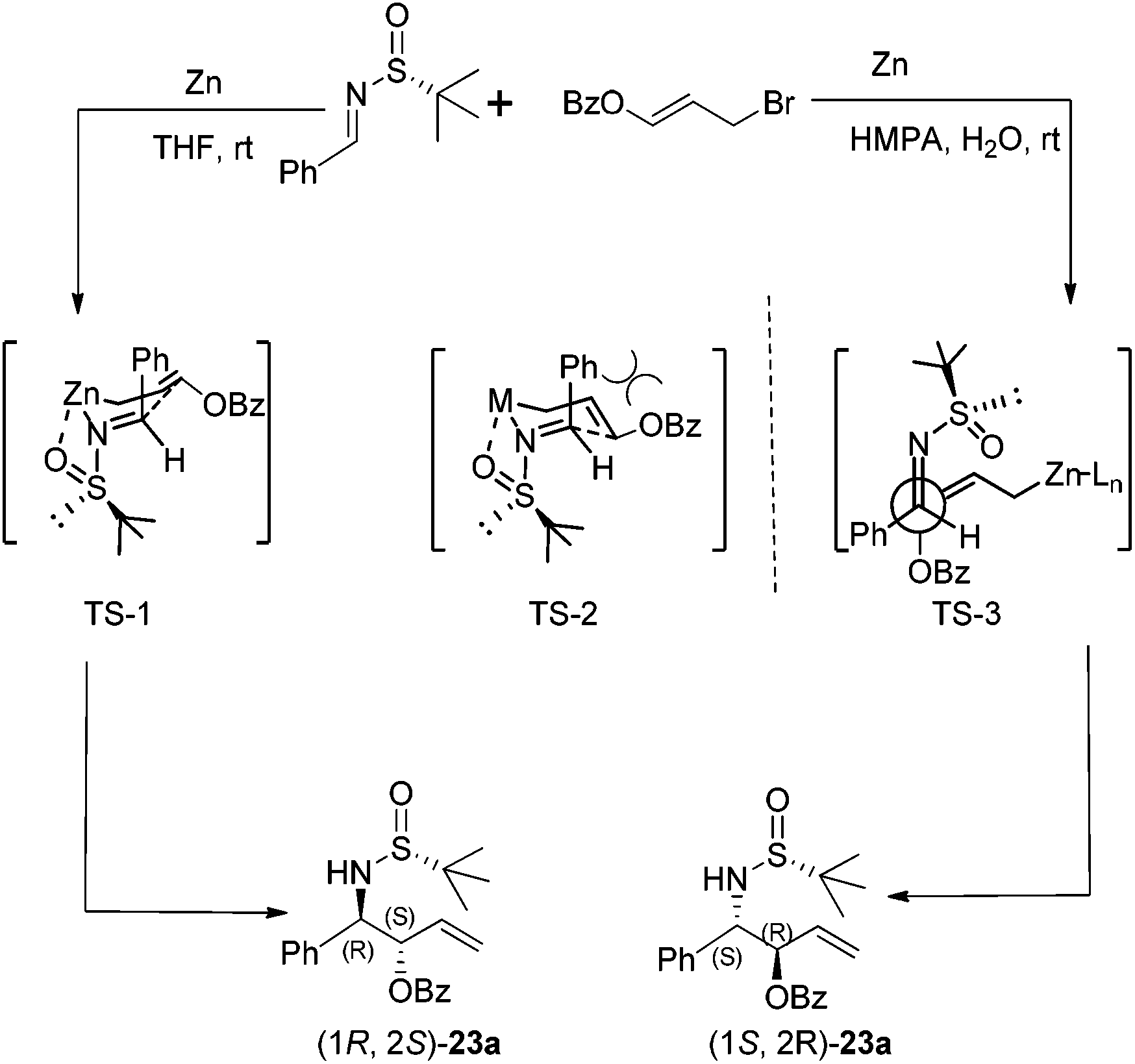 | ||
| Scheme 9 Proposed reaction pathways for Zn-mediated benzoyloxyallylation. | ||
The synthetic application of the above benzoylallylation was demonstrated by the efficient asymmetric synthesis of (−)-cytoxazone, a selective modulator of Th2 cytokine secretion isolated from Streptomyces sp.26 The allylation of p-methoxybenzaldehyde derived (S)-imine with 3-benzoyloxyallyl bromide could provide the corresponding anti-product 24 in almost quantitative yields, with a 98:2 anti/syn ratio and 92% de. The basic hydrolysis of benzoate and acidic cleavage of the chiral sulfinyl group resulted in β-amino alcohol, which was treated with 1,1′-carbonyldiimidazole (CDI) to give oxazolidinone 25 in 89% overall yield. Ozonolysis of the double bond and its subsequent reduction with sodium borohydride accomplished the total synthesis of (−)-cytoxazone (Scheme 10).
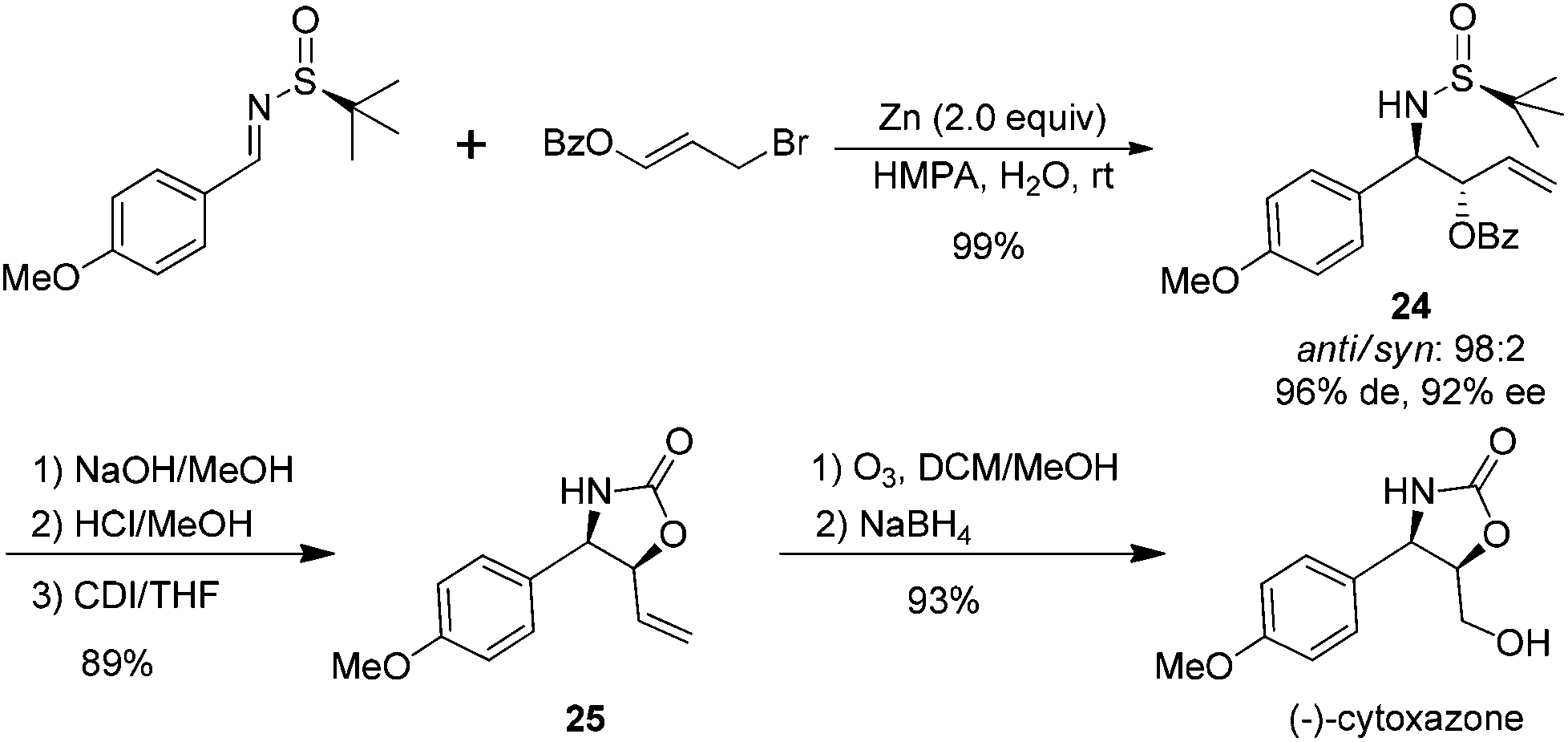 | ||
| Scheme 10 Total synthesis of (−)-cytoxazone through asymmetric benzoyloxyallylation. | ||
The reaction of N-tert-butanesulfinyl imines with 3-arylallyl bromide (cinnamyl bromide) 26 could lead to various β-aryl homoallylic amines 27.24 A dramatic lithium chloride effect on the stereocontrol of this reaction was observed after a careful study.27 Under the optimal conditions, the imines were treated with 2 equivalents of zinc, 2 equivalents of (E)-1-bromo-4-(3-bromoprop-1-enyl)benzene and 1 equivalent of LiCl in DMF, which was distilled from activated Al2O3, providing the corresponding homoallylic amines with a syn/anti ratio up to 99:1 and diastereoselectivity up to 99% de (Scheme 11). It is noteworthy that the use of absolute anhydrous DMF, which was distilled from CaH2, could cause an obvious decrease in stereoselectivity (with syn/anti ratio of 90:10 and 92% de). The cinnamylation of a series of (R)-N-tert-butanesulfinyl imines was investigated. For aromatic imines, the nature of aryl and heteroaryl R groups did not have a major impact on the syn/anti ratios (95:5 to 99:1), as well as the diastereoselectivities (95–99% de). The reactions with imines derived from aliphatic aldehydes, such as cinnamaldehyde, 3-phenylpropanal and 3-methylbutanal, also proceeded well, giving the corresponding syn-products 27 in 95–97% de. Interestingly, the reaction with either (E) or (Z)-cinnamyl bromide almost exclusively afforded the syn-isomer, with syn/anti ratios as 97:3 and 98:2 and diastereoselectivities as 99% and 97% de, respectively.
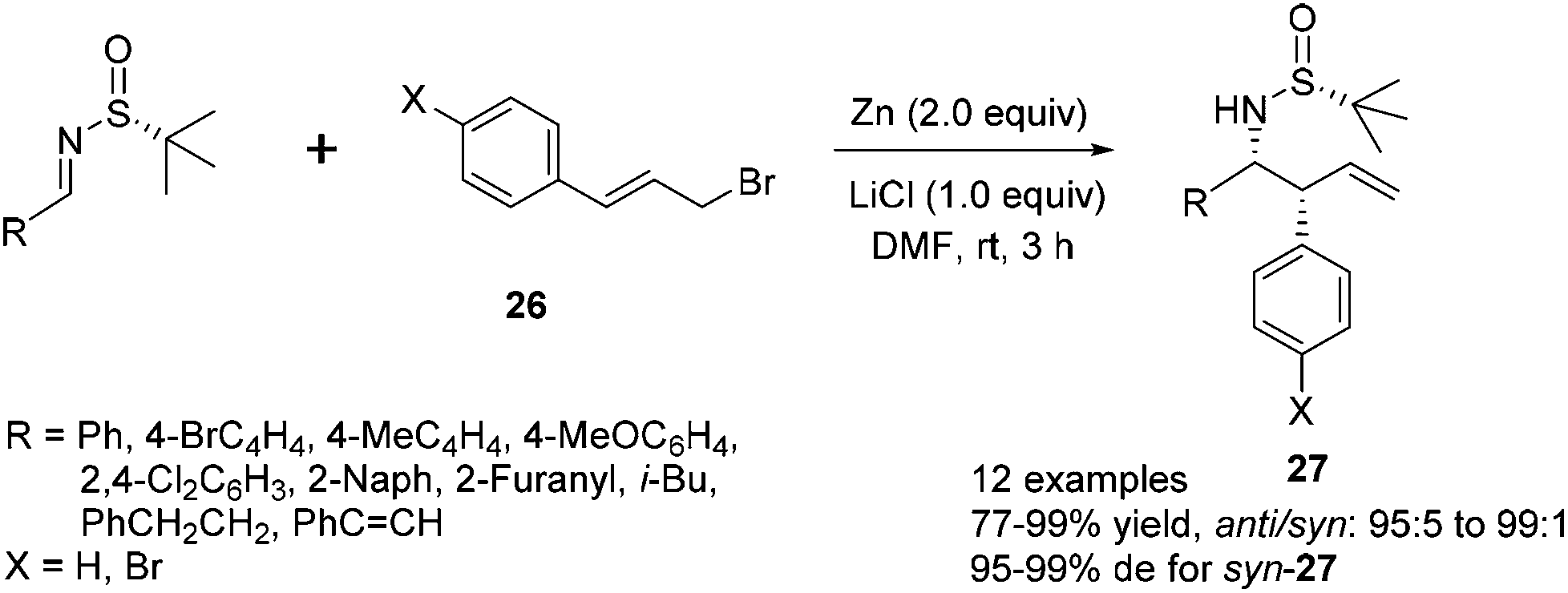 | ||
| Scheme 11 Zn-mediated cinnamylation to provide β-aryl homoallylic amines. | ||
An attractive application of this cinnamylation method was to use N-sulfinyl α-imino ester 12 as an electrophilic substrate (Scheme 12). In this case, the reactions could provide chiral α-amino acid derivatives with a β-vinyl moiety, in higher yields and with better stereoselectivities without the additive LiCl.
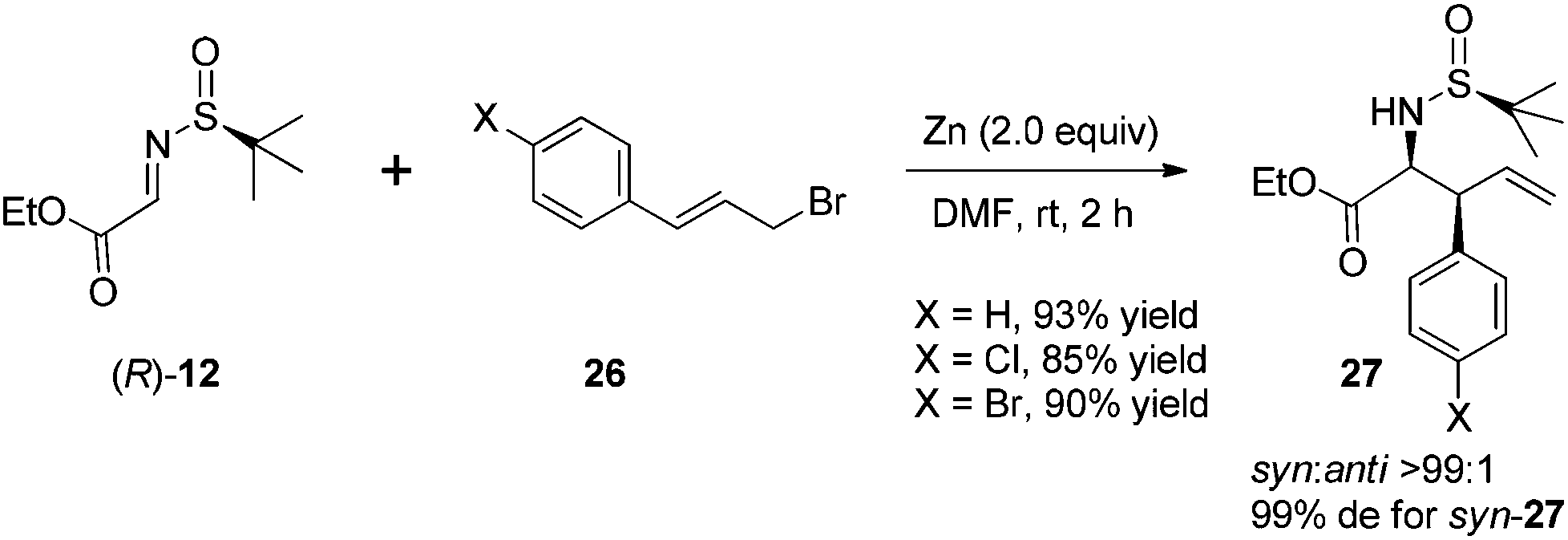 | ||
| Scheme 12 Preparation of β-vinyl-containing α-amino acid derivatives. | ||
The reaction of N-tert-butanesulfinyl imines with 3-methyl-2-ethoxycarbonylallylic bromide (ethyl 2-bromomethyl butenoate) 29 could result in highly substituted γ-lactams bearing an α-methylene group (Scheme 13).28a Under the optimal conditions, the imines were treated with 3 equivalents of zinc, 3 equivalents of (Z)-ethyl 2-(bromomethyl) butenoate and 5 equivalents of LiCl in DMF, followed by the cleavage of the chiral auxiliary with 1 N HCl in dioxane. This one-pot process could produce highly substituted α-methylene-γ-lactams in good yields (51–89%) with excellent trans/cis ratios (97.1:2.9 to 99.7:0.3 when R′ = Me) and enantio-purities (92–99% ee). Various aromatic imines, including aryl and heteroaryl substitutions, afforded the corresponding products with excellent enantioselectivities and diastereoselectivities. The imines derived from aliphatic aldehydes, such as 3-phenylpropanal, 3-methylbutanal, also gave excellent stereoselectivities although only moderate isolated yields were obtained. The synthetic application was demonstrated by the total synthesis of tubulysin V.28b As a complementary study for such a one-pot asymmetric synthesis of poly-substituted trans-α-methylene-γ-lactams, we achieved the highly efficient construction of cis-α-methylene-γ-lactams via the Zn-mediated allylation of N-tert-butanesulfinyl using 3-(bromomethyl)coumarin as the precursor of an allylic reagent.28c
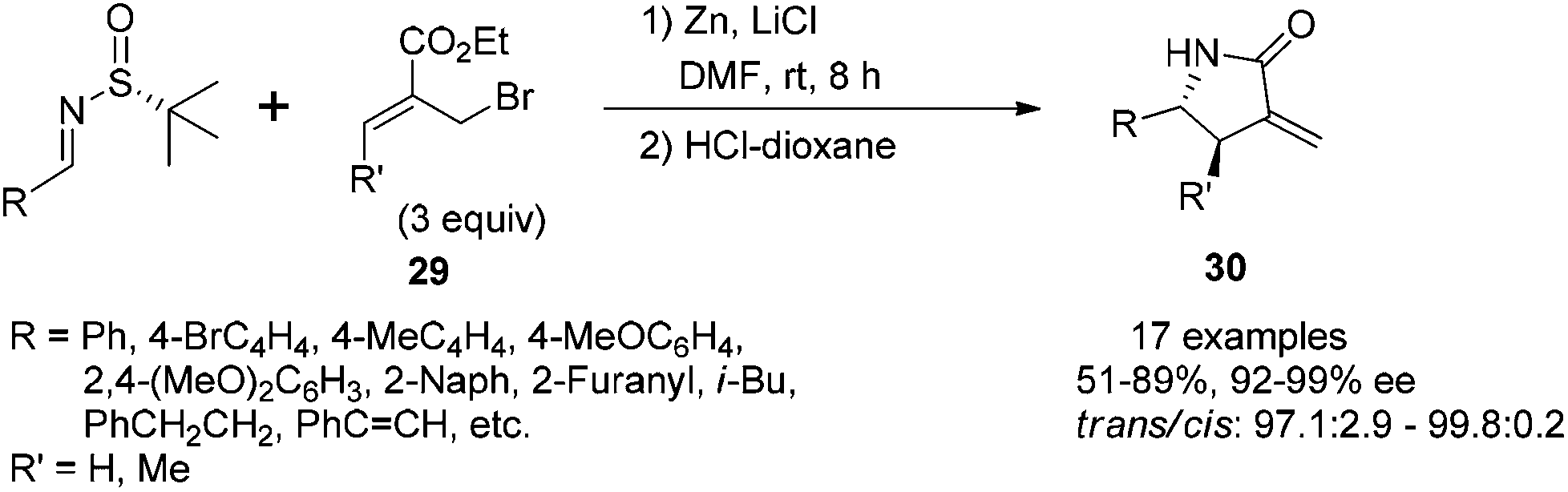 | ||
| Scheme 13 One-pot preparation of highly substituted α-methylene γ-lactams. | ||
The successful application of N-tert-butanesulfinyl imines in the reaction with various linear allylic bromides, including 3-bromopropenyl benzoate, cinnamyl bromide and ethyl 2-bromomethyl butenoate, prompted us to explore the opportunity with heterocyclic allylic bromide. As shown in Scheme 14, 3-bromomethyl-5H-furan-2-one 31 could react with N-tert-butanesulfinyl imines, providing optically pure α-methylene-γ-butyrolactones bearing chiral amino moieties in the β-position.29 Under the optimal conditions, the imines were treated with 3 equivalents of zinc and 3 equivalents of 3-bromomethyl-5H-furan-2-one in DMF, along with 2 equivalents of water, which likely played an important role to protonate the zinc salt of the allylation adduct. The use of excessive water led to a decrease in the reaction yield, due to the decomposition of the allylic zinc reagent. The reactions produced a mixture of anti- and syn-isomers, which could be separated by silica gel flash chromatography. For the aryl and alkenyl imines, the reactions afforded the major anti-isomers with excellent diastereoselectivities (generally >99% de). However, the diastereoselectivity of the reaction with alkyl imine (derived from 3-phenylpropanal) significantly dropped to 76% de, due to the reduced steric hindrance of the alkyl substituent. Notably, the resulting α,β-unsaturated lactone skeleton from this asymmetric allylation could allow for further rhodium-catalyzed arylation, which introduced a new chiral center in a stereospecific manner (Scheme 15).
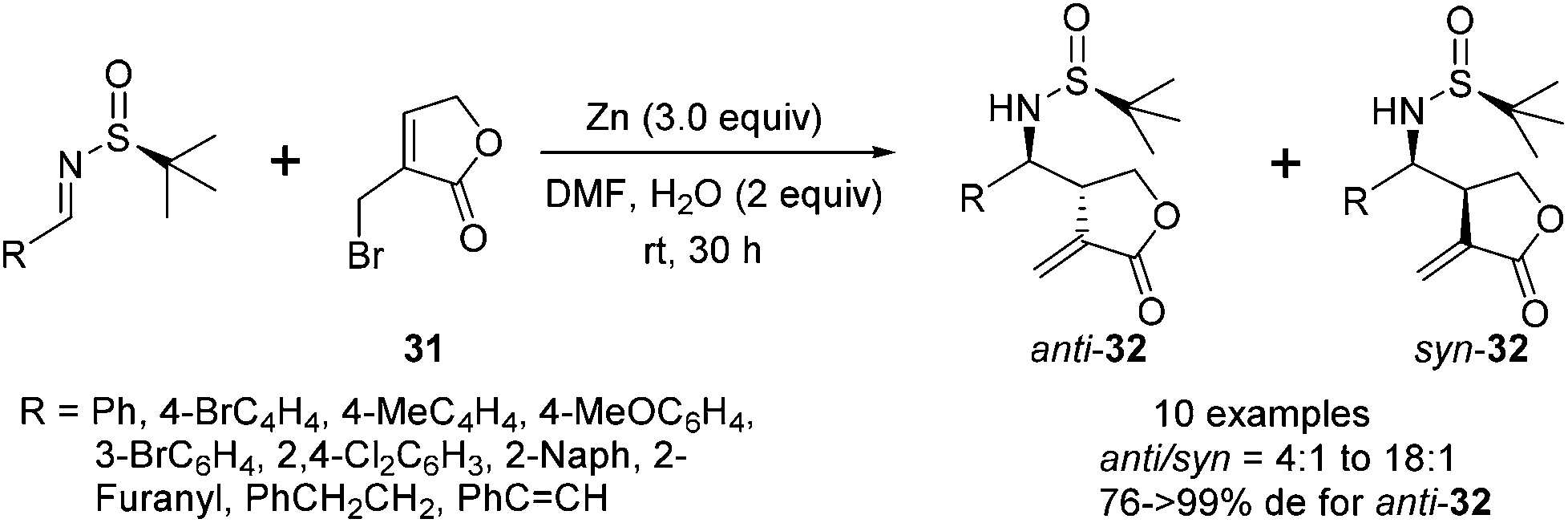 | ||
| Scheme 14 Zn-mediated allylation of N-tert-butanesulfinyl imines using 3-bromomethyl-5H-furan-2-one. | ||
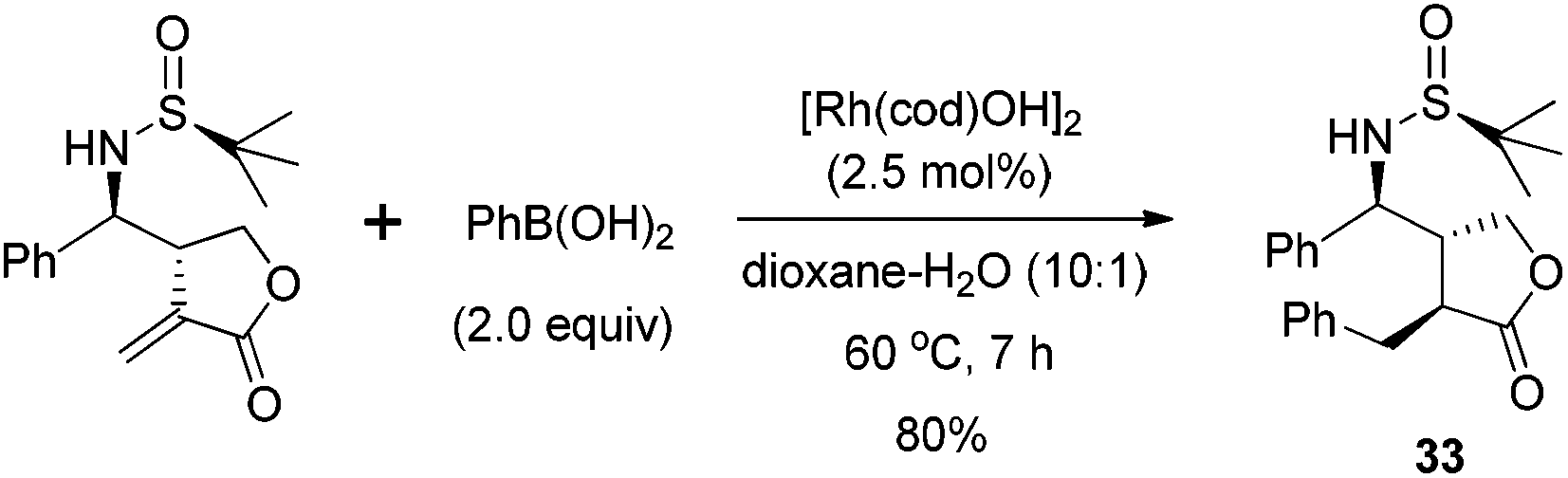 | ||
| Scheme 15 Rhodium-catalyzed arylation of anti-isomer to introduce another chiral center. | ||
In addition to these successful applications of various allylic bromide substrates, we recently conducted a systematic research on the allylation of chiral N-tert-butanesulfinyl ketimines, which were scarcely studied due to their relatively lower reactivity. Although the steric hindrance difference in the two sides of ketimines became less significant, moderate to excellent enantioselectivities were still achieved for this Zn-mediated allylation using THF as the solvent (Scheme 16). A six-membered transition-state chair model was proposed for the stereochemical outcome in this case.30
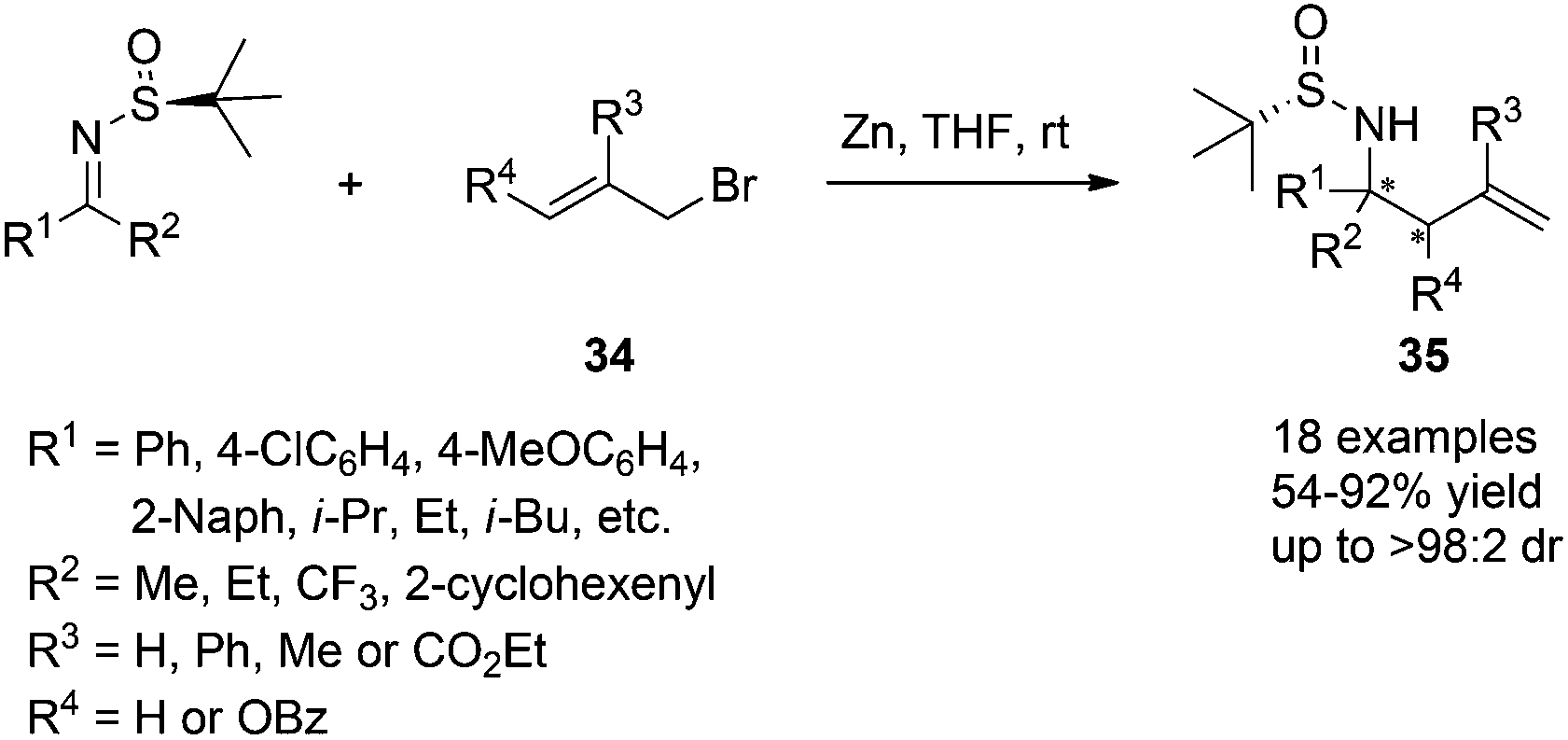 | ||
| Scheme 16 Zn-mediated allylation of N-tert-butanesulfinyl ketimines. | ||
The zinc-mediated diastereoselective allylation of isatin-derived N-tert-butanesulfinyl ketimines was also achieved at room temperature, providing a highly practical asymmetric approach for the efficient synthesis of chiral tetra-substituted 3-aminooxindoles (Scheme 17).31
 | ||
| Scheme 17 Zn-mediated allylation of N-tert-butanesulfinyl ketimines. | ||
3. Synthetic applications of chiral diene ligands
Another part of the research focus in our group is to discover new catalytic transformations based on chiral diene ligands. While pursuing the above carbon–carbon bond formation through aza Barbier-type allylation of N-tert-butanesulfinyl imines, we further explored the synthetic application of our unique diene ligands highlighted in Fig. 4. The nonbridged bicyclo[3.3.0]octadiene-based ligands were independently introduced by our group32 and Laschat group33 in 2007. Both enantiomers in bicyclo[3.3.0]octadiene series could be prepared from the commercially available cycloocta-1,5-diene, with lipase-catalyzed enzymatic resolution as a key step. Later, a new family of chiral diend ligand based on the dicyclopentadiene skeleton was also reported by our group.34 The synthesis of C1-symmetric dicyclopentadiene-based ligands applied a similar synthetic sequence using commercial dicyclopentadiene as the starting material. In both cases, the kinetic resolution through lipase-catalyzed acetylation could lead to both enantiomers in >98% ee. Recently, the chiral bicyclo[3.3.0]octadiene ligand was successfully incorporated into a polymer chain. Such immobilized chiral diene ligands demonstrated similar catalytic activity and could be recycled for multiple uses.35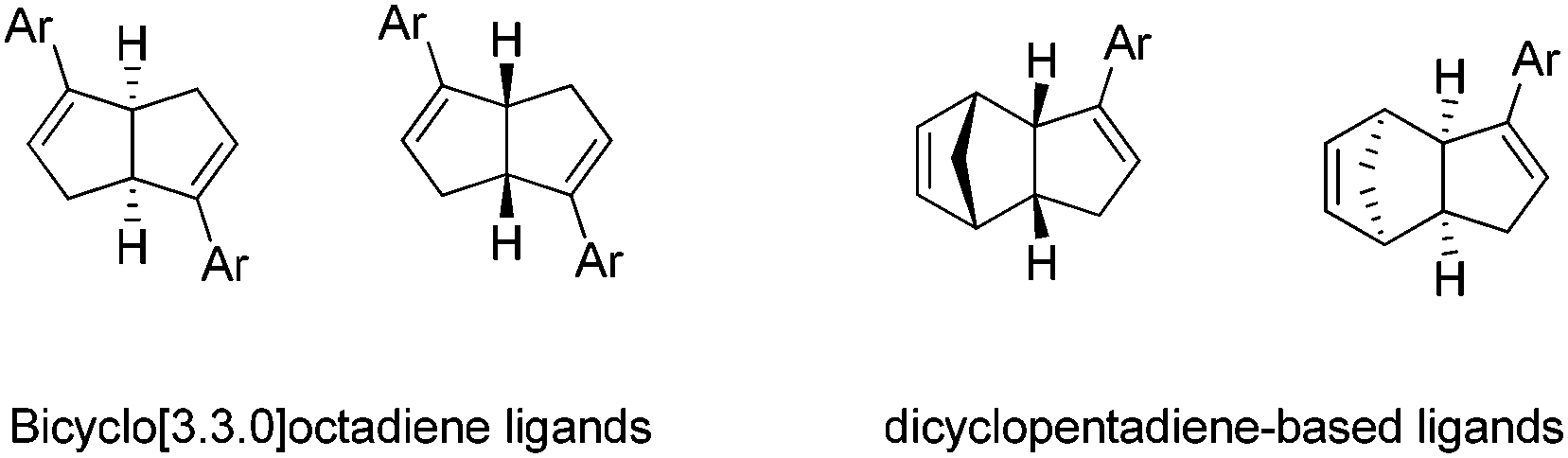 | ||
| Fig. 4 Our representative chiral diene ligands. | ||
3.1. Rhodium–diene catalyzed asymmetric addition to imines
Transition metal-catalyzed asymmetric reaction, including the design of efficient chiral ligands, is always an attractive topic in organic synthesis. Since the pioneering work on chiral diene ligands by Hayashi36 and Carreira,37 various diene ligands have been designed and evaluated in asymmetric catalysis, especially in rhodium- and iridium-catalyzed asymmetric reactions. In many cases, the diene ligands demonstrated better stereoselectivity and catalytic activity than the powerful phosphine ligands.38 Our diene ligands in both series were successfully utilized in rhodium-catalyzed enantioselective arylation of N-tosyl imines with aryl boronic acids, and further extended to rhodium-catalyzed conjugate addition of α,β-unsaturated ketones/esters, nitroalkenes, and α,β-unsaturated γ-lactams. In addition to various rhodium-catalyzed reactions, we successfully demonstrated the synthetic utility of chiral palladium–diene complexes in the Suzuki–Miyaura cross-coupling reaction.Our first synthetic application of C2-symmetric bicyclo[3.3.0]octadiene was the rhodium-catalyzed arylation of aryl N-tosyl imines with aryl boronic acids.1c,39 The reaction was carried out with 2 equivalents of aryl boronic acid in the presence of 2 equivalents of triethylamine, 3 mol% of [RhCl(C2H4)2]2 and 3.3 mol% of chiral diene at 55 °C for 4–5 h (Scheme 18). A variety of aryl N-tosyl imines, regardless of their steric and electronic effects, could afford the corresponding diarylmethyltosylamides 41 with extremely high enantioselectivities (98–99% ee). In addition to the substituted phenyl imines, other aromatic imines including furanyl, thienyl, naphthyl proved to be excellent substrates for this transformation. An attractive application of this asymmetric addition was to use 2-methoxycarbonyl phenyl imine 42 as the reaction substrate, which could conveniently generate chiral phthalimidines (isoindolones) 43 in good yields and excellent enantioselectivities (Scheme 19).30 Bicyclic heterocyclic imines, for example, N-tosyl and N-nosyl indolylimines, could also react with aryl boronic acids, providing a new enantioselective approach for the synthesis of α-aryl 2- or 3-indolyl-methanamines.40
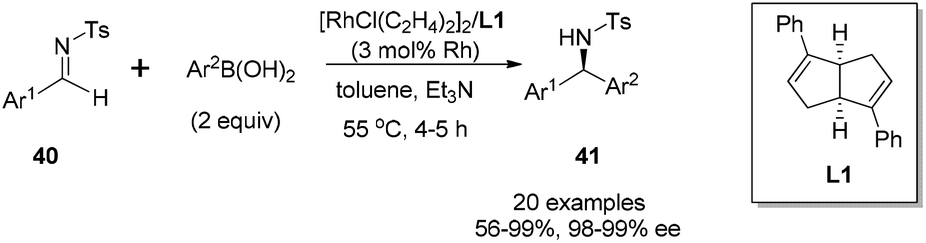 | ||
| Scheme 18 Catalytic asymmetric arylation of aryl N-tosyl imines with arylboronic acids. | ||
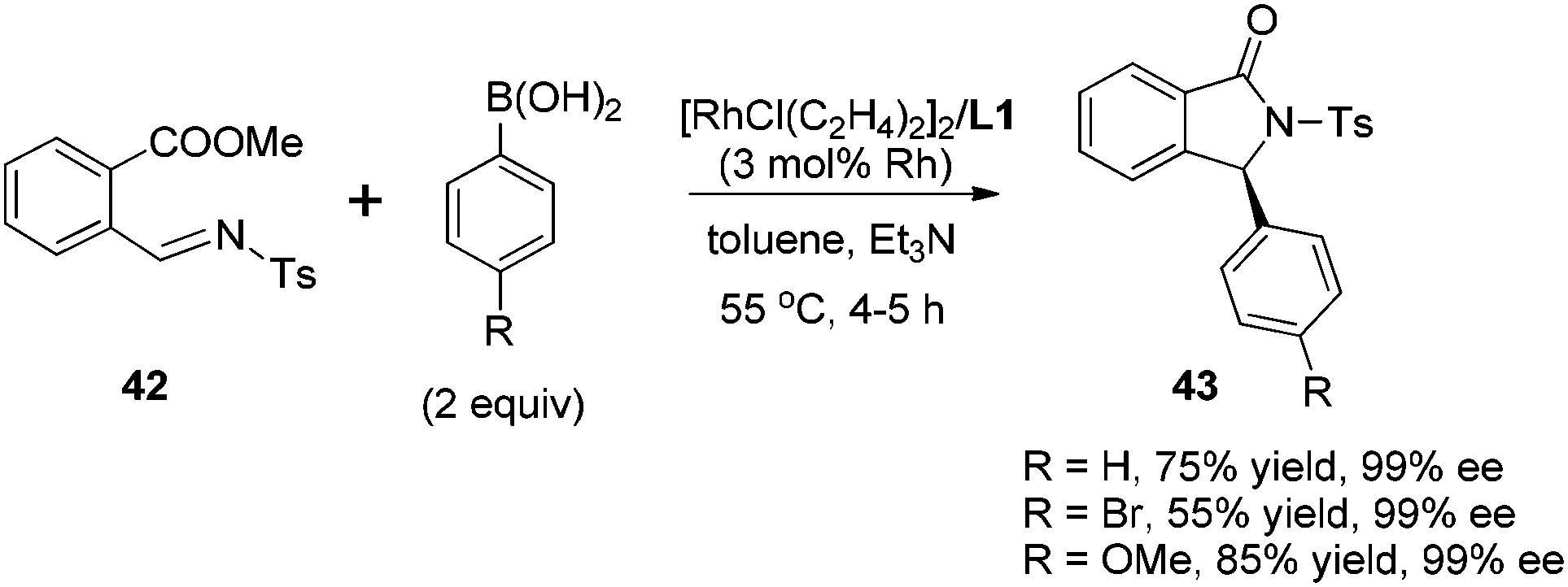 | ||
| Scheme 19 Synthesis of chiral 3-aryl-2-tosyl-2,3-dihydroisoindolones. | ||
The arylation of aliphatic imines is much more challenging due to the low stability of the imines.41 Although the arylation of aliphatic N-tosyl imines could also yield the corresponding adducts under the above conditions in excellent enantioselectivity (99% ee), the isolated yield was quite low (only 28% yield for imine substrate derived from propanal). Attempts to improve the reaction yield by switching the chiral diene ligand, as well as the additive, turned out to be fruitless. Thus a more reactive catalyst [Rh(OH)(L)]2 and a neutral reaction condition were applied. This replacement significantly improved the arylation of N-tosylimine of propanal, providing the desired products in almost quantitative yields (99%), along with excellent enantioselectivity (>99% ee).42 Under the optimal conditions, various aliphatic N-tosyl imines were treated with 2 equivalents of aryl boronic acids, rhodium–diene complex [Rh(OH)(L)]2 (3 mol% Rh) and 4 Å molecular sieves in dioxane, affording the corresponding chiral sulfonamides 45 in high yields (87–99%) and with excellent enantioselectivities (91–>99% ee) (Scheme 20). Similar results were also achieved for N-nosylimine substrates.
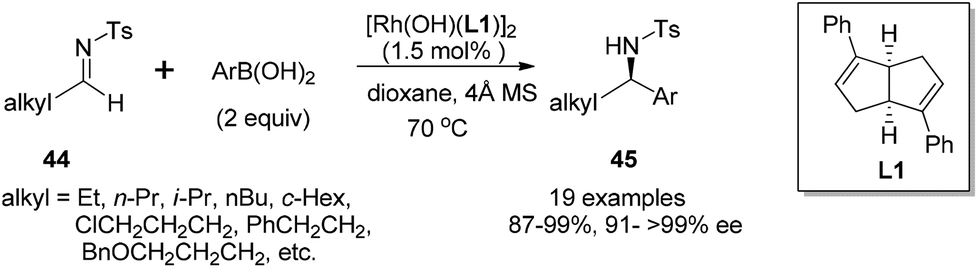 | ||
| Scheme 20 Catalytic asymmetric arylation of aliphatic N-tosyl imines with arylboronic acids. | ||
It is noteworthy that the imines with terminal functional groups such as Ph, BnO, and Cl were also suitable substrates, giving the desired adducts in 87–99% yields and >99% ee. The products with terminal Cl substitution could be further cyclized upon treatment with potassium carbonate to form chiral 2-aryl pyrrolidine and piperidine derivatives 47 in a one-pot fashion (Scheme 21), and the N-tosyl group in the cyclized products could be cleaved through the reduction with naphthalene/Li, affording useful optically pure 2-aryl pyrrolidine and piperidine building blocks.
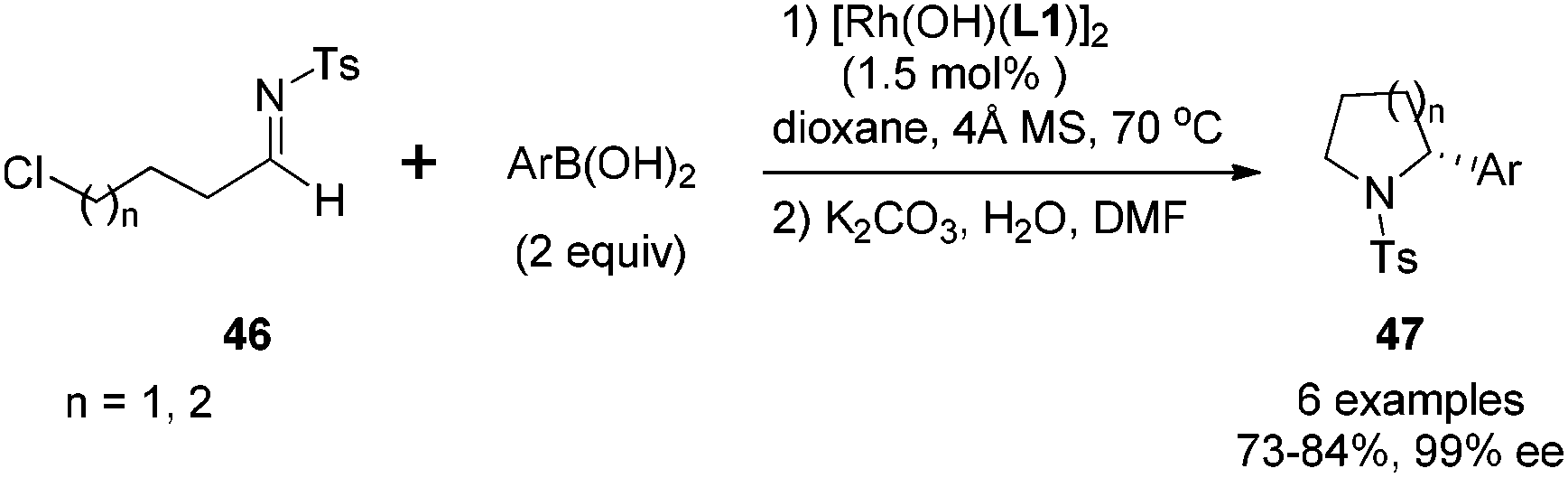 | ||
| Scheme 21 One-pot synthesis of chiral 2-aryl pyrrolidine and piperidine derivatives. | ||
The C1-symmetric chiral dicyclopentadiene-based ligands were also applied in the rhodium-catalyzed arylation of aryl N-tosyl imines (Scheme 22).43 In this case, the additive of aqueous KHF2 provided higher yields and better enantioselectivities than the use of Et3N as a base. Compared with the arylation using bicyclo[3.3.0]octadiene ligands, this new series led to the corresponding diarylmethyltosylamides 41 in higher yields (98–99%), but with slightly lower enantioselectivities (90–96% ee).
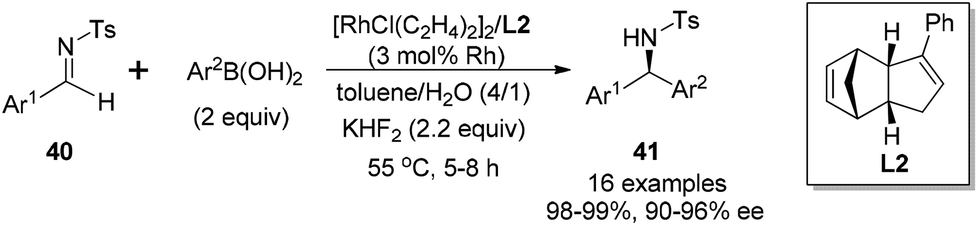 | ||
| Scheme 22 Catalytic asymmetric arylation of aryl N-tosyl imines with arylboronic acids. | ||
In addition to these extensive studies on the arylation of imines, we also investigated the rhodium catalyzed alkenylation of arylimines. Due to the relatively lower stability of alkenylboronates and the possible competitive binding between chiral diene and the alkenylation reagent/product, this reaction would be much more challenging. Lam and co-workers reported the alkenylation of stable and relatively reactive cyclic imines.44 The only single successful example of acyclic imines was provided by Shintani, Hayashi and co-workers during their research on the arylation of imines.45 Our studies provide a more general approach toward this end. A variety of potassium alkenyltrifluoroborates were successfully added to various N-tosyl arylimines, providing the corresponding products in high yields and excellent enantioselectivies (Scheme 23). The power of this method was demonstrated by the short formal synthesis of the natural product (−)-aurantioclavine (Scheme 24).46
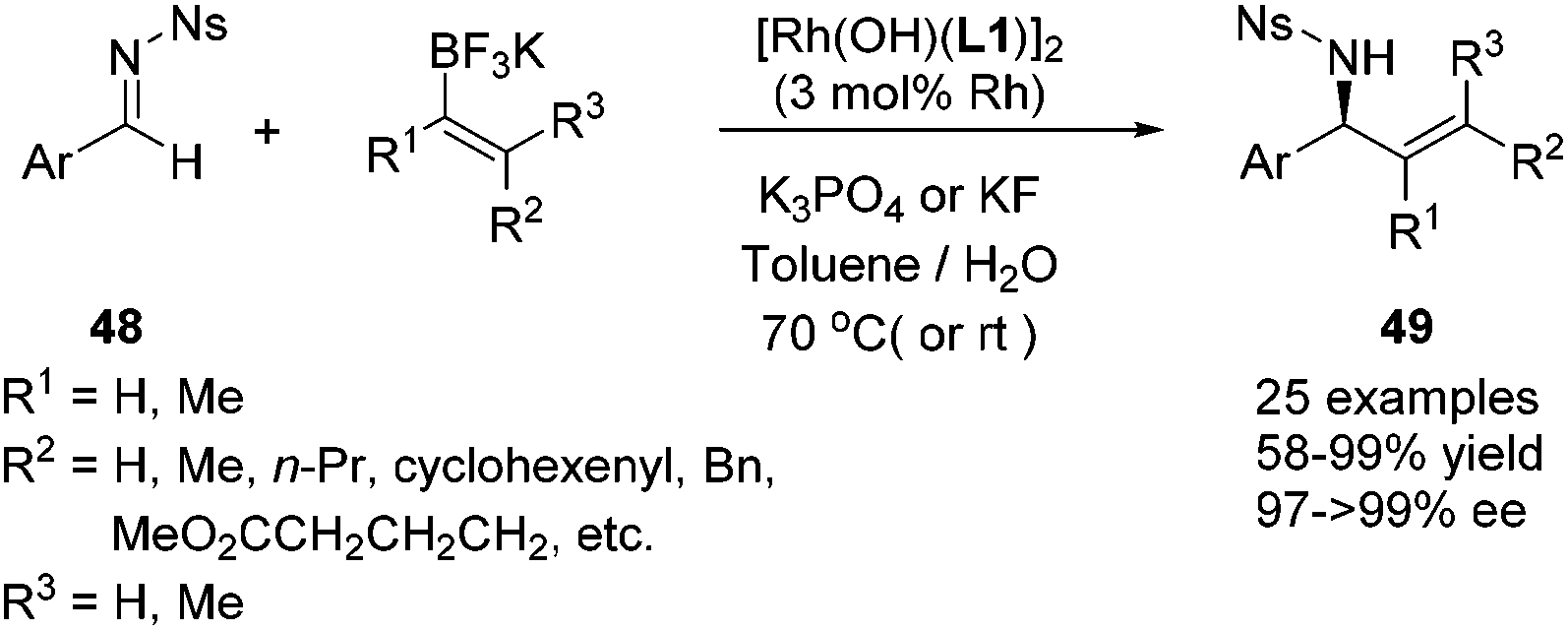 | ||
| Scheme 23 Catalytic asymmetric alkenylation of aryl N-nosyl imines with potassium alkenyltrifluoroborates. | ||
 | ||
| Scheme 24 Formal synthesis of (−)-aurantioclavine. | ||
3.2. Rhodium–diene catalyzed asymmetric conjugate additions
In addition to the rhodium-catalyzed asymmetric arylation to aliphatic and aromatic imines, the chiral diene ligands also demonstrated excellent activity and stereoselectivity in the rhodium-catalyzed asymmetric conjugate additions to α,β-unsaturated ketones/esters, nitroalkenes, and α,β-unsaturated γ-lactams.As shown in Scheme 25, a series of aryl boronic acids could react with 2-cyclopentenone (X = CH2, n = 1), 2-cyclohexenone (X = CH2, n = 2), and cyclic α,β-unsaturated esters (X = O, n = 1, 2). The reactions proceeded under very mild conditions with 2 equivalents of boronic acid, 2.5 mol% of [RhCl(C2H4)2]2 and 5.5 mol% of chiral diene ligand at room temperature for 3 h, providing the corresponding chiral adducts 54 in excellent yields and enantioselectivities.47 In this case, the electron-donating or electron-withdrawing substitution of the phenyl boronic acids did not significantly affect the reaction yields and stereoselectivities. Interestingly, sterically more hindered 1-naphthylboronic acid and 2-tolylboronic acid led to slightly higher enantioselectivities (98% ee) for this conjugate addition.
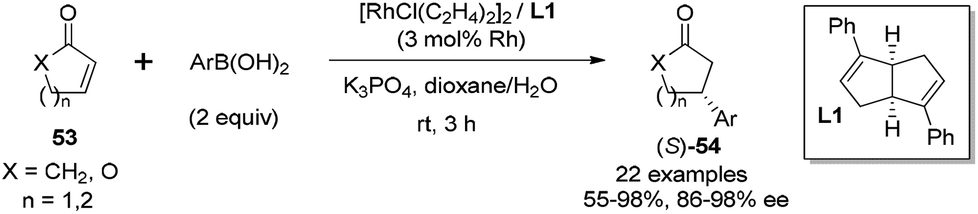 | ||
| Scheme 25 Catalytic asymmetric conjugate addition with cyclic α,β-unsaturated ketones/esters. | ||
The above bicyclo[3.3.0]octadiene ligand has two phenyl rings directly connected to two double bonds. It would be interesting to explore other ligands L3–5 by moving the bulky substitutions from the double bonds to the adjacent carbon atoms. Due to multiple oxygen atom substitutions, such diene ligands are shown to be quite soluble in water (Fig. 5).
 | ||
| Fig. 5 Solubility of chiral diene ligands in water. | ||
We tried rhodium-catalyzed asymmetric conjugate addition of cyclohexenone and phenyl boronic acid using the above ligands in aqueous media. The reactions were conducted with 2 equivalents of boronic acid, 2.5 mol% of [RhCl(C2H4)2]2, 5.5 mol% of chiral diene ligand at room temperature. To our delight, all three ligands L3–5 could lead to the corresponding adduct within 0.5 h, and the diketal ligand L3 was found to be optimal, providing high yield (96%) and excellent enantioselectivity (93% ee).48 Encouraged by this, we explored such addition reaction with a variety of boronic acids (Scheme 26). Different from the results in Scheme 25, the 2-cyclohexenone (X = CH2, n = 2) substrate afforded higher enantioselectivities than the 2-cyclopentenone (X = CH2, n = 1) substrate, providing the corresponding adducts 54 with 93–95% ee and 80–86% ee, respectively. In addition, the sterically more hindered 1-naphthylboronic acid and 2-methoxyphenylboronic acid resulted in significant loss in terms of stereoselectivities (52% ee and 69% ee, respectively). The reactions with cyclic α,β-unsaturated ester (X = O, n = 2) also led to a modest decrease in enantioselectivities (80–82% ee).
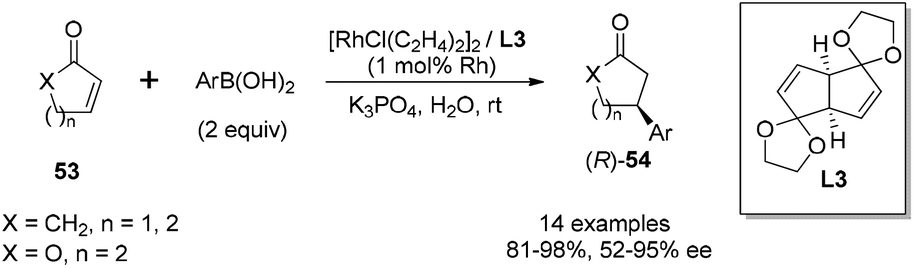 | ||
| Scheme 26 Catalytic asymmetric conjugate addition with cyclic α,β-unsaturated ketones/esters. | ||
In contrast to the ineffectiveness of diphenyl diene ligand L1 for the linear α,β-unsaturated substrates, this hydrophilic diketal diene ligand L3 could promote such asymmetric conjugate additions to linear α,β-unsaturated ketones (Scheme 27). This transformation was also very sensitive to the steric factor of boronic acids, but in the opposite direction. Sterically encumbered arylboronic acids, such as 2-chlorophenylboronic, 2-tolylboronic and 1-naphthylboronic acids, proved to be favorable for higher enantioselectivities. To the best of our knowledge, this serves as the first example of an asymmetric reaction employing a chiral metal–diene catalyst in water.
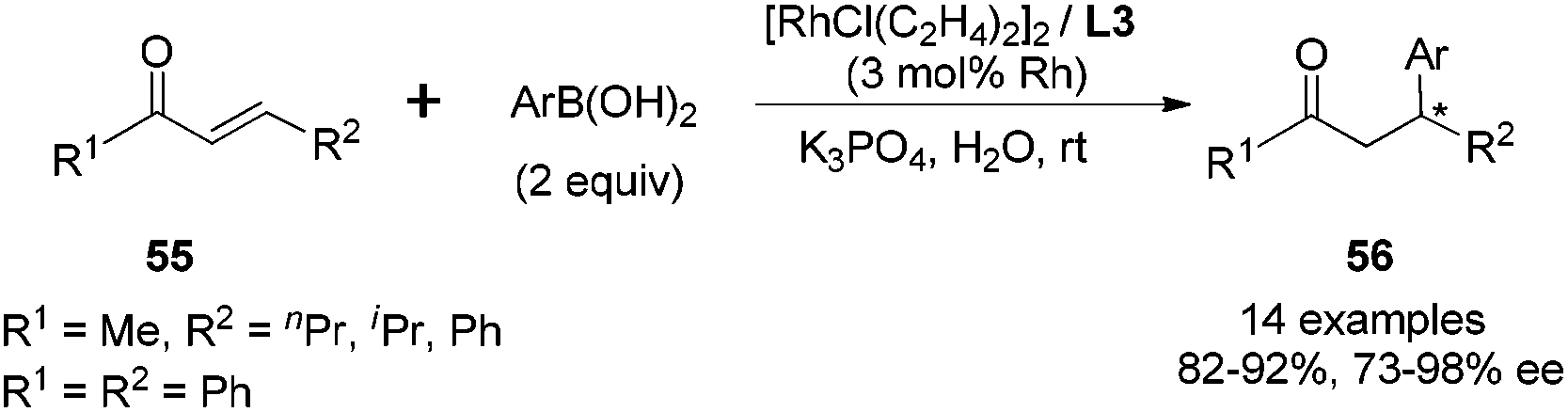 | ||
| Scheme 27 Catalytic asymmetric conjugate addition with linear α,β-unsaturated ketones. | ||
The chiral dicyclopentadiene ligands could also induce excellent enantioselectivities for the rhodium-catalyzed asymmetric conjugate addition.49 As shown in Scheme 28, both cyclic and linear substrates could undergo this asymmetric addition, affording the corresponding adducts. In general, the 2-cyclohexenone (X = CH2, n = 2) substrate afforded higher enantioselectivities (usually >96% ee) than 2-cyclopentenone and a linear α,β-unsaturated ketone. The electronic properties of the substitution groups at the phenyl ring of arylboronic acids did not affect the yields and enantioselectivities. However, the sterically hindered boronic acids, such as 2-methoxyphenylboronic and 1-naphthylboronic acid, led to a modest decrease in stereoselectivity, giving the corresponding adducts with 87% ee and 85% ee, respectively. As for the linear enone substrates, 2-methylphenylboronic acid afforded much higher enantiomeric excess (87% ee) than phenylboronic acid did (16% ee) although both reactions were accomplished in high yields (90–92%).
 | ||
| Scheme 28 Catalytic asymmetric conjugate addition with α,β-unsaturated ketones/esters. | ||
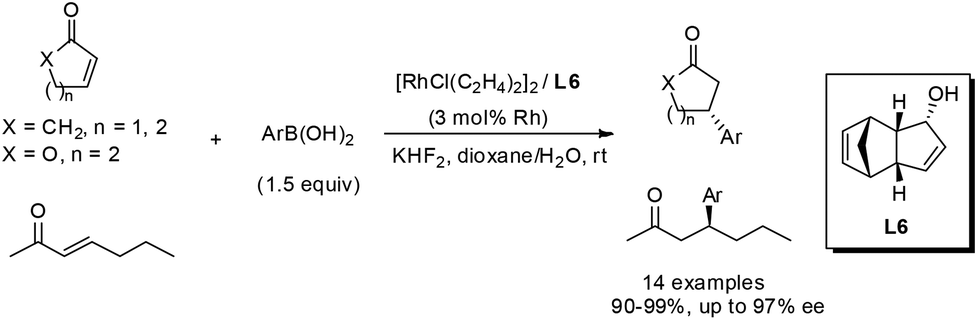
The rhodium-catalyzed asymmetric conjugate addition of boronic acids to nitroalkenes was also demonstrated with the chiral bicyclo[3.3.0] diene ligands.50 In this case, the addition of 3 equivalents of KHF2 proved to be crucial for high yields and enantioselectivities, and the reaction was performed in toluene–water at 100 °C (Scheme 29). As for the aromatic substituted nitroalkenes, the sterically hindered aryl boronic acids, such as 1-naphthylboronic and 2-tolylboronic acid, tended to afford higher enantioselectivities, giving the corresponding adducts 58 with 95% ee and 97% ee, respectively. The electron-withdrawing group at the phenyl ring of boronic acids could enhance the selectivity. In contrast, the electronic properties of the substitution at the phenyl ring of nitroalkene substrates 57 did not significantly affect the reaction selectivities. Interestingly, the aliphatic nitroalkene (R = cyclohexyl) and nitrostyrene (R = Ph) could give similar enantioselectivities (78% ee and 84% ee, respectively) when reacting with 4-anisylboronic acid. The reaction with linear 1-nitrohexene (R = n-Bu) led to lower enantioselectivity (61% ee), but the selectivity could get enhanced to 86% ee when 4-methoxyphenylboronic acid was replaced by sterically encumbered 2-tolylboronic acid. It is worth highlighting that our results represent the best enantioselectivity in the asymmetric addition of arylboronic acids to α-unsubstituted nitroalkene substrates.
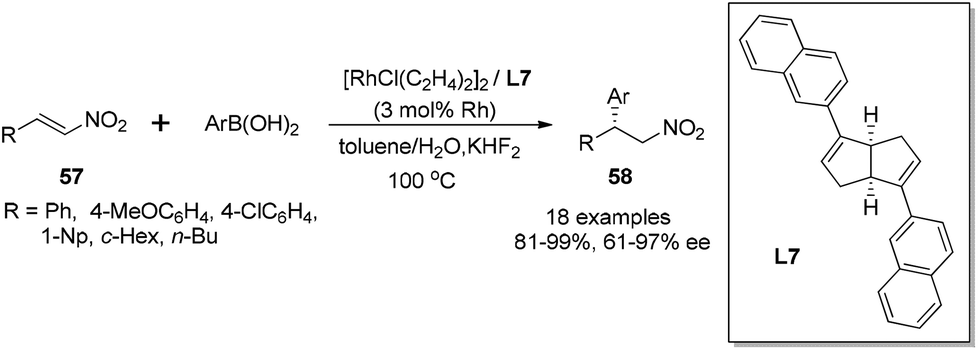 | ||
| Scheme 29 Catalytic asymmetric conjugate addition with nitroalkenes. | ||
The synthetic utility of this highly selective asymmetric addition was exemplified by the following preparation of isoquinoline derivative (Scheme 30). The corresponding chiral adduct 59 from 4-fluorophenyl boronic acid could be conveniently converted, through the Bischler–Napieralski cyclization, to obtain optically active (S)-4-(4-fluorophenyl)-6,7-dimethoxy-3,4-dihydroquinoline 61 in three steps.
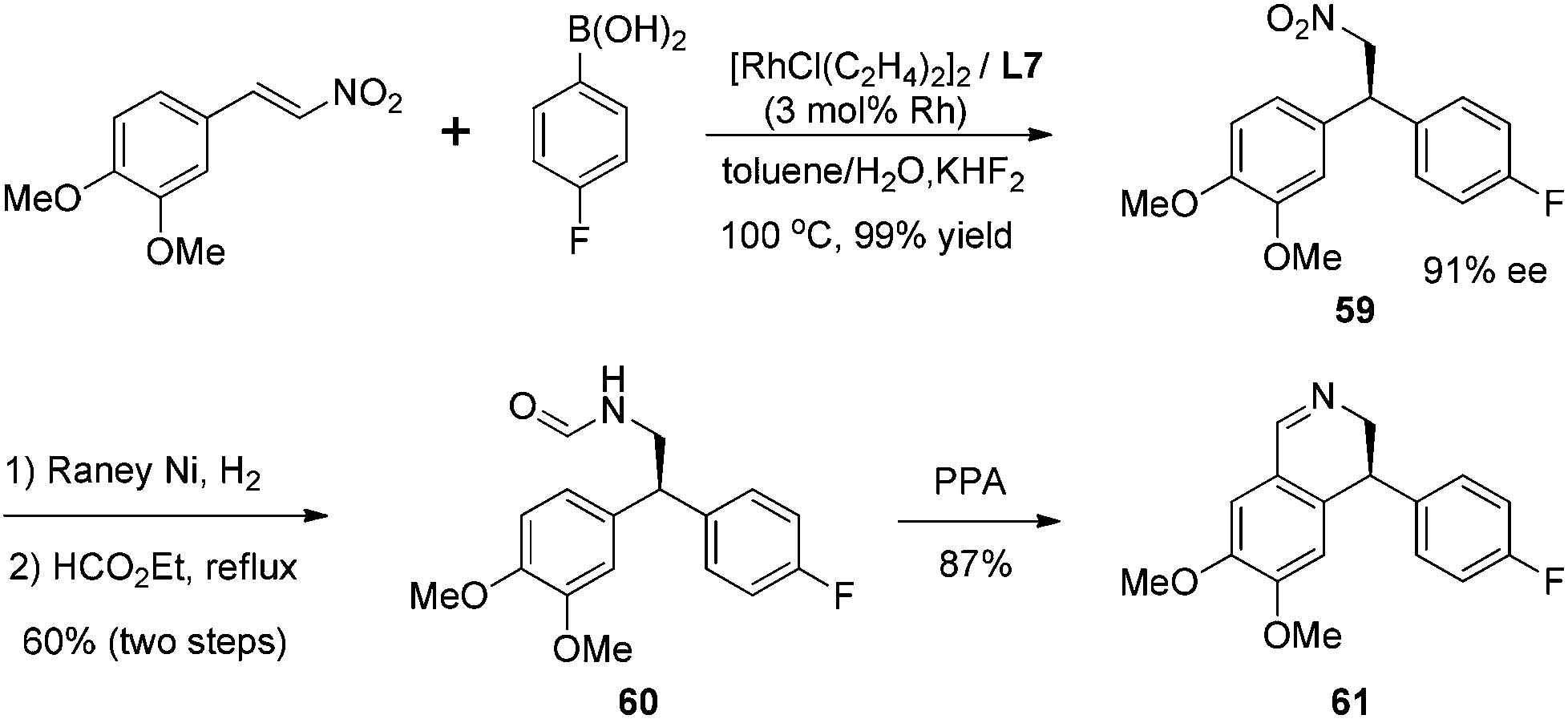 | ||
| Scheme 30 Synthesis of isoquinoline derivative. | ||
Having established the rhodium-catalyzed asymmetric conjugate addition for α,β-unsaturated ketones/esters and nitroalkenes, we turned our focus to investigate α,β-unsaturated γ-lactam substrates.51 The studies showed that the N-Boc protection was crucial for this conjugate addition because protection with either benzyl or the 4-methoxyphenyl group led to inseparable alkene by-products, which were generated through Mizoroki–Heck type coupling with aryl boronic acids. As shown in Scheme 31, a series of arylboronic acids successfully reacted with the N-Boc protected α,β-unsaturated γ-lactam 62 under optimal conditions, providing the corresponding adducts 63 in high yields and with excellent enantioselectivities (97–99% ee). The steric properties of arylboronic acids had little effect on the performance of the addition. However, electron-withdrawing groups (4-CF3, 4-Cl, 4-Br) at the phenyl ring of boronic acids led to a significant drop in the reaction yields in spite of excellent enantioselectivities. Fortunately, this issue was solved by switching the additive from Et3N to KHF2, which could dramatically shorten the reaction time and thus improved the yields from 56–78% to >97%. This transformation provided a facile access of chiral γ-lactams, which could be applied in the synthesis of the GABAB receptor agonist (R)-baclofen and the antidepressant (R)-rolipram.
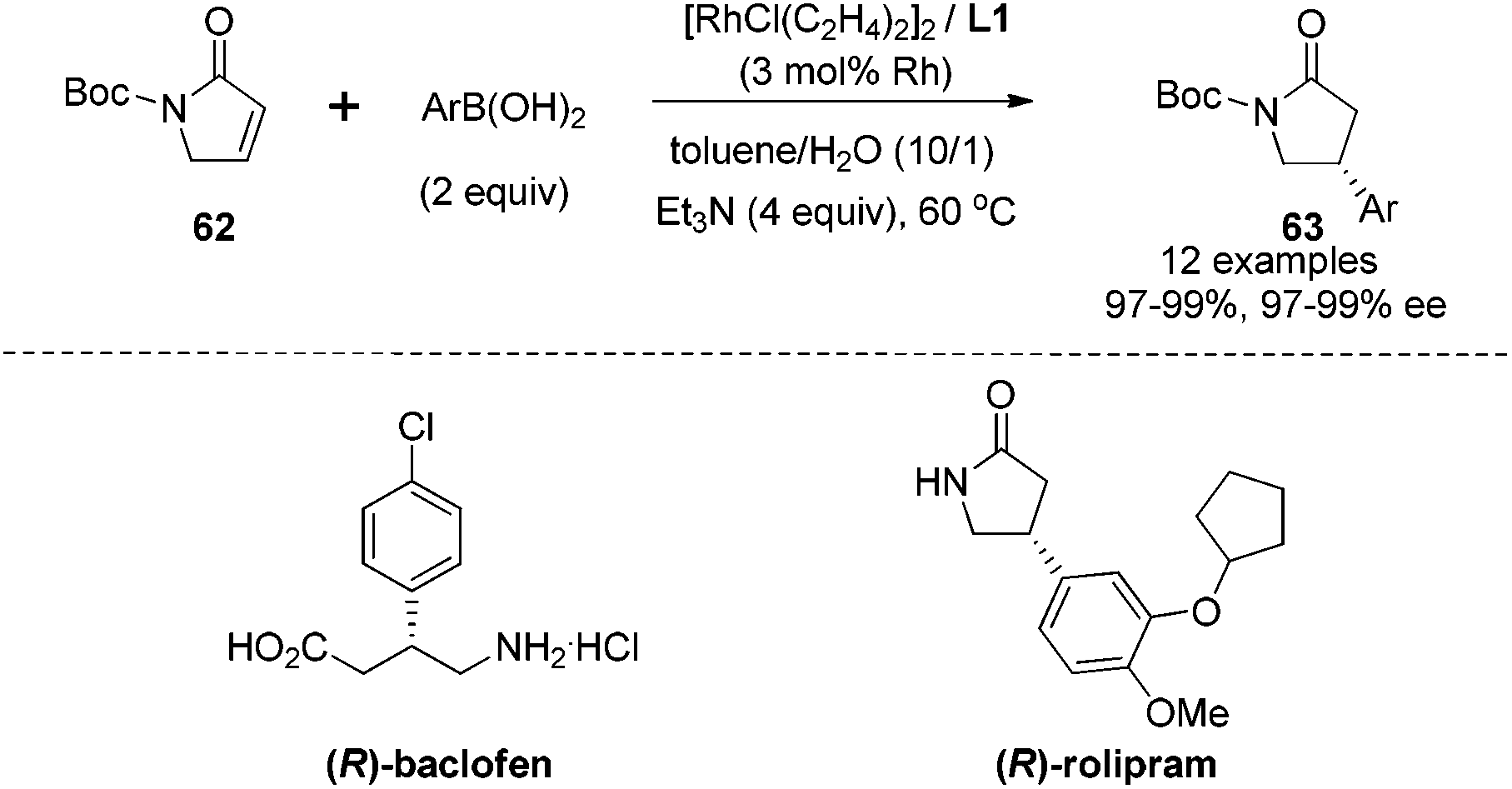 | ||
| Scheme 31 Catalytic asymmetric conjugate addition with α,β-unsaturated γ-lactam. | ||
Compared with arylboron reagents, the use of alkenylboron reagents is much less examined for the rhodium-catalyzed asymmetric conjugate addition.52 Although many groups reported the addition of alkenylboron reagents to simple α,β-unsaturated ketones,53 their addition to α,β-unsaturated amides remains unexplored. Encouraged by the performance of our diene ligands in the conjugate addition to N-Boc protected α,β-unsaturated γ-lactam, we turned our focus to extend arylboron reagents to alkenylboron reagents. Due to the low stability of alkenylboronic acids, we chose to apply potassium alkenyltrifluoroborates in the rhodium-catalyzed conjugate addition (Scheme 32).54 For the γ-lactam substrate 64 (n = 1), the reactions with a variety of alkenyltrifluoroborates all afforded excellent enantioselectivities (97–99% ee), but the reaction yields were dependent on both steric and electronic properties of alkenyltrifluoroborates. The reactions with more substituted alkenyltrifluoroborates or electron-rich alkenyltrifluoroborates generally led to higher yields. For the δ-lactam substrate 64 (n = 2), the enantioselectivities were slightly lower (91–94% ee) while the yields remained high. In addition to the reactions with α,β-unsaturated lactams, alkenyltrifluoroborates could react with cyclic α,β-unsaturated ketones and esters 66. In the case of the cyclic α,β-unsaturated esters (lactones), the combination of dioxane and KF turned out to be better than toluene and Et3N system.
 | ||
| Scheme 32 Catalytic asymmetric conjugate addition with alkenyltrifluoroborates. | ||
The synthetic application of this asymmetric transformation was demonstrated by the total synthesis of (−)-α-kainic acid, an alkaloid natural product first isolated in 1953 from the Japanese marine algae Digenea simplex.55 As shown in Scheme 33, the reaction of γ-lactam with potassium 1-methylvinyltrifluoroborate, by the catalysis of the rhodium-(R,R)-diene complex, afforded the desired conjugate adduct 68 in 82% yield and with 99% ee. Its subsequent alkylation exclusively gave the trans-isomer 69, which was isomerized to cis-isomer 70 through a thermodynamic protonation process. Further manipulations of the amide functional group, through the reaction of an aminal intermediate 71 with TMSCN and a subsequent basic hydrolysis, completed the total synthesis of (−)-α-kainic acid.
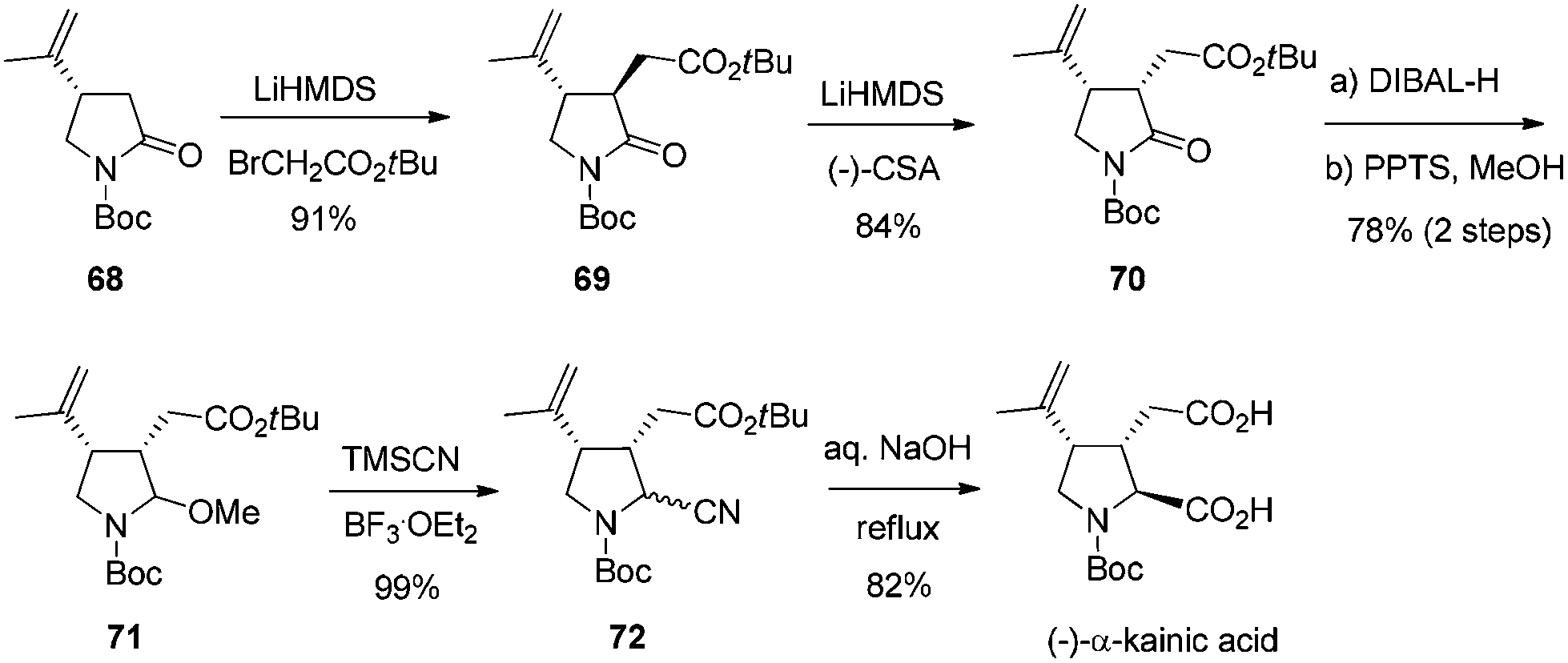 | ||
| Scheme 33 Synthesis of (−)-α-kainic acid. | ||
3.3. Palladium–diene catalyzed asymmetric Suzuki–Miyaura cross-coupling reactions
Although diene ligands are widely applied in rhodium- and iridium-catalyzed asymmetric reactions, there were very few examples of the use of chiral diene ligands in palladium-catalyzed asymmetric transformations. In 2005, a chiral diene–palladium complex was prepared and tested in enyne cyclization reaction. The desired product was generated, but in a racemic form and with low yield (35%).56 The application of a palladium–diene complex in asymmetric reactions faces the following two major challenges: first, palladium-catalyzed reactions often require the change of metal valence state in the catalytic cycle, which is different from the rhodium-catalyzed transformations where the metal valence state usually keeps constant. Second, the diene ligands probably possess weaker binding affinity than phosphorus ligands to stabilize palladium in different oxidation states. With significant efforts in further expanding the synthetic application of our diene ligands, we found that bicyclo[3.3.0]octadiene ligands could form air-stable complexes with Pd(II) by treatment with PdCl2(PhCN)2 in benzene at room temperature overnight. From the screening of different ligands for Suzuki–Miyaura cross-coupling, the 3,5-dimethylphenyl group substituted bicyclo[3.3.0]octadiene was found to provide the corresponding axially chiral biaryls in the combination of good yields and enantioselectivities (Scheme 34).57 Under the optimal conditions, a variety of aryl bromides and trifluoromethanesulfonates could react with aryl boronic acids in the presence of Pd–diene complex 75 (5 mol%) and a free ligand (15 mol%) using 2.5 equivalents of Cs2CO3 as a base in toluene. It is noteworthy that some reactions, for example, the coupling between 2-bromo-3-methylbenzaldehyde and 2-methylphenyl boronic acid, could occur at 0 °C to provide the corresponding biaryl in 97% yield and with 65% ee. To the best of our knowledge, this coupling reaction serves as the first successful application of chiral diene ligands in the asymmetric palladium-catalyzed transformation.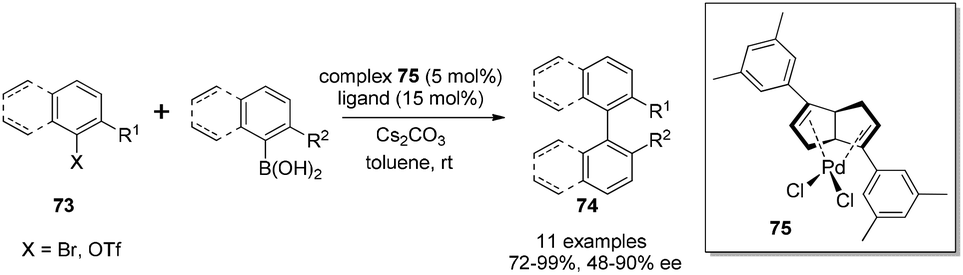 | ||
| Scheme 34 Catalytic asymmetric Suzuki–Miyaura cross-coupling. | ||
X-ray crystallography of two Pd–diene complexes was obtained by coordination with diphenyl- and di(3,5-dimethylphenyl)-substituted dienes. A structural analysis confirms that our cis-fused bicyclic diene skeleton creates a wedge shape, providing an excellent chiral environment to coordinate with PdCl2, which is quite similar to the Rh–diene complexes.41 A detailed comparison of the bond lengths and angles between free dienes and the corresponding Pd-complexes in the X-ray crystallography was conducted through a Dewar–Chatt–Duncanson model. The length of double bond in the complexes (1.37–1.38 Å) is longer than that in the corresponding free ligand (1.32 Å), indicating an sp3 characteristic after binding with PdCl2. In addition, the dihedral angle of the bicyclic diene unit shrinks (88–91° versus 107°) and creates a bite angle (85–86°) after binding with palladium. The dihedral angle in di(3,5-dimethylphenyl)-substituted diene complex (87.6°) is slightly smaller than that in the diphenyl-substituted diene complex (90.8°), indicating that the di(3,5-dimethylphenyl)-substituted diene results in stronger chelating affinity with PdCl2. The mechanism of this palladium–diene catalyzed asymmetric Suzuki–Miyaura cross-coupling was investigated using electrospray ionization mass spectrometry (ESI-MS/MS).58 In all detected palladium species, the diene ligand was firmly associated with palladium, explaining the high enantioselectivities for this asymmetric transformation.
4. Development of chiral sulfur–olefin ligands for asymmetric reactions
The allylation of chiral N-tert-butanesulfinyl imines could generate a variety of enantiopure N-tert-butanesulfinyl homoallylic amines bearing different substitution patterns at the C-1 and C-2 positions. The chiral products from this asymmetric allylation actually could serve as attractive olefin–sulfur hybrid ligands.59 As shown in Scheme 35, Xu observed that a simple chiral N-tert-butanesulfinyl homoallylic amine L8 led to an excellent conversion for the rhodium-catalyzed asymmetric arylation, affording the desired conjugate adduct in 99% yield and with 82% ee. The modification of either of the functional groups involved in the ligand binding with rhodium, i.e., the reduction of olefin or elimination of sulfur chirality, resulted in detrimental effects on this asymmetric transformation. The former (compound L9) did not generate any desired product, while the latter (compound L10) afforded the desired product in a racemic form and with much lower yield (25%). This observation indicates that the sulfinamide and allylic double bond are both involved in the coordination with rhodium during the catalytic process. In addition, the N-methyl analog (compound L11) resulted in very poor conversion, probably due to the unfavorable coordination geometry. This study represents one of the first examples using chiral sulfur–olefin ligands for transition metal-catalyzed asymmetric transformation.60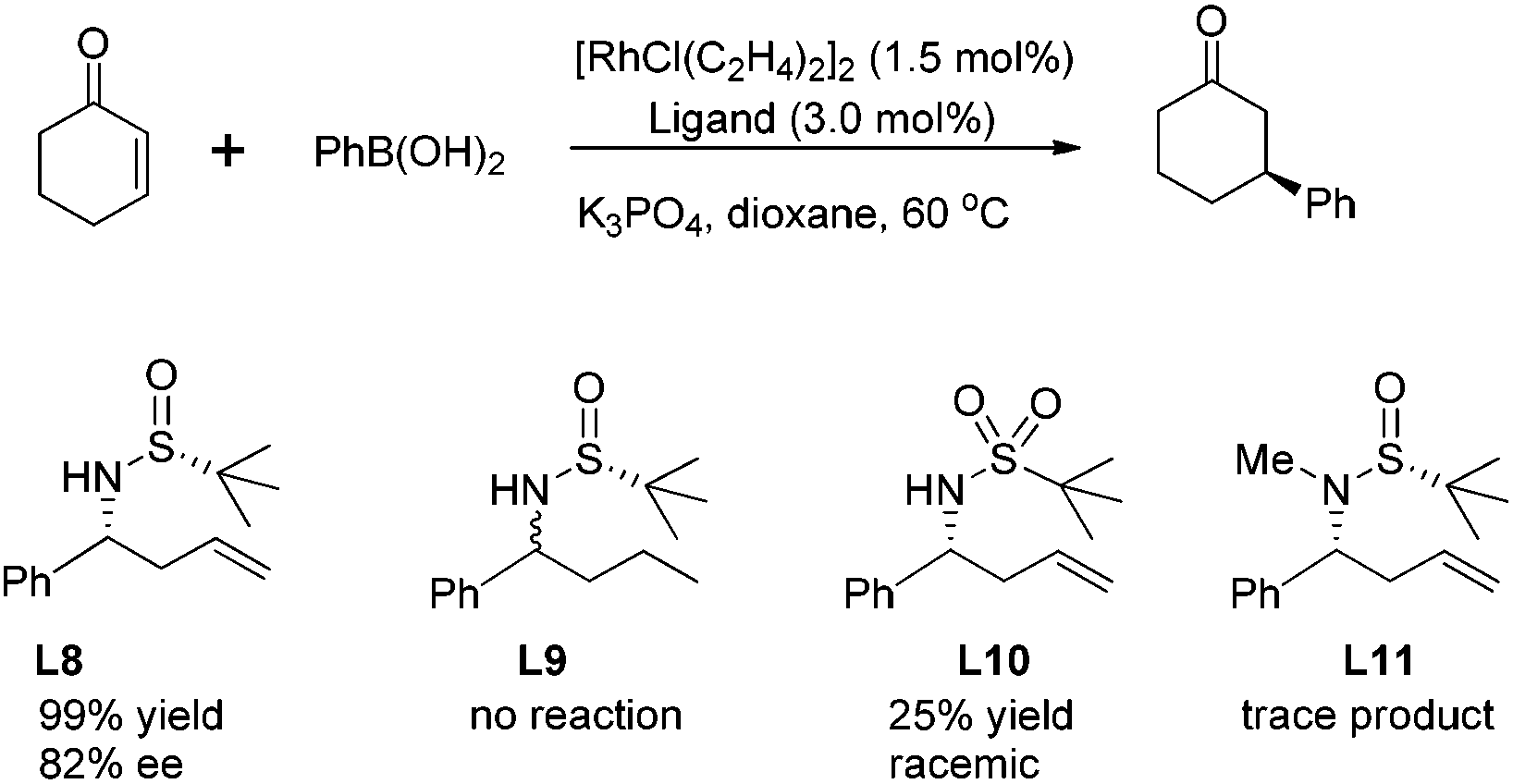 | ||
| Scheme 35 Proof-of-concept studies on potential N-tert-butanesulfinyl homoallylic amine as ligand. | ||
With the encouraging results, we evaluated a series of N-tert-butanesulfinyl homoallylic amines accumulated over the last decade as chiral ligands for this rhodium-catalyzed conjugate addition (Fig. 6). Except the ligand L19, which gave quite a poor yield (21%) and low enantioselectivity (−26% ee), other ligands promoted the asymmetric arylation in almost quantitative yields (99%) and with excellent enantioselectivities (82–95% ee). Interestingly, both diastereomers L8 and L12, regardless of the absolute configuration of the carbon chiral center, afforded the addition product displaying the same chirality with similar enantioselectivities (82% and 85% ee, respectively). The benzoates L13–18, which were generated by the asymmetric benzoyloxyallylation, all promoted the conjugate addition in high yields (99%) and with excellent enantioselectivities (94–95% ee). It is noteworthy that both the anti-isomers L17 and L18 led to the same chiral adduct in almost quantitative yields (99%) and with outstanding enantioselectivities (95% and 94% ee, respectively). A separate experiment with a mixture of two anti-isomers as ligands also demonstrated a highly enantioselective formation of the addition product in 99% yield and with 95% ee. In other words, the enantioselectivity of this conjugate arylation was mainly controlled by the stereochemistry of the chiral N-sulfinyl group. This observation provides great advantage for ligands L17 and L18 in the rhodium-catalyzed asymmetric arylation as it can simply apply the products from zinc-mediated benzoyloxyallylation of the corresponding chiral N-tert-butanesulfinyl imine regardless of its stereoselectivity outcome.
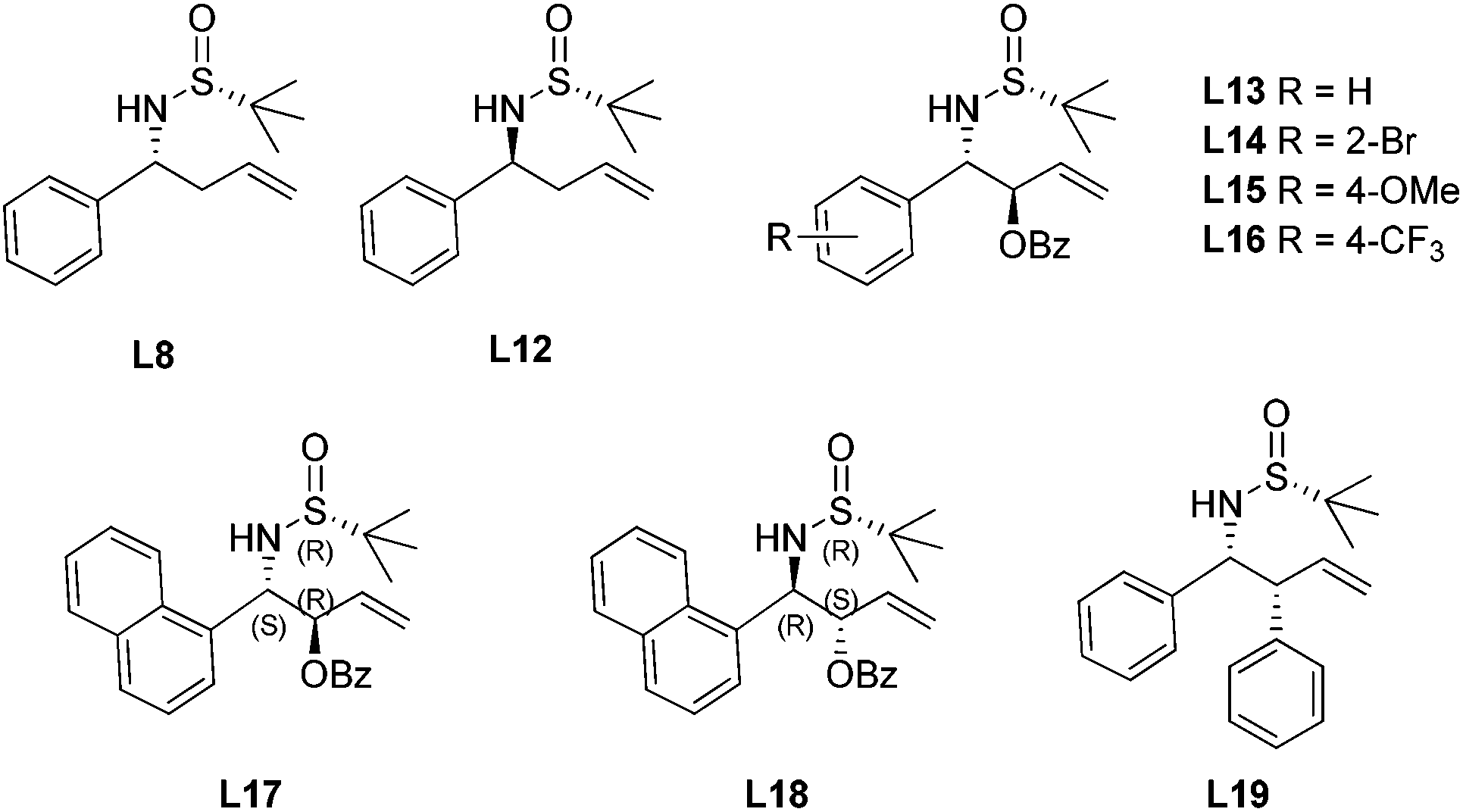 | ||
| Fig. 6 Sulfinamide–olefin ligands. | ||
With the simple and readily available chiral ligand L18, different substrates for the rhodium-catalyzed asymmetric arylation were examined. A variety of α,β-unsaturated cyclic carbonyl compounds, including 2-cyclohexenone, 2-cyclopentenone, α,β-unsaturated δ-lactone and lactam, could undergo the 1,4-addition, affording the corresponding products in high yields (74–99%) and with excellent enantioselectivities (78–98% ee) (Scheme 36). The steric and electronic properties of aryl boronic acids appeared to have little influence on the reaction. Unfortunately, the reaction of linear enone (E)-2-heptenone with phenyl boronic acid gave the desired product in low yield (51%) and stereoselectivity (38% ee). Our continuous work in this field revealed that a simpler ligand L21 also showed great catalytic activity and enantioselectivity for the conjugate addition to α,β-unsaturated cyclic carbonyl compounds.61
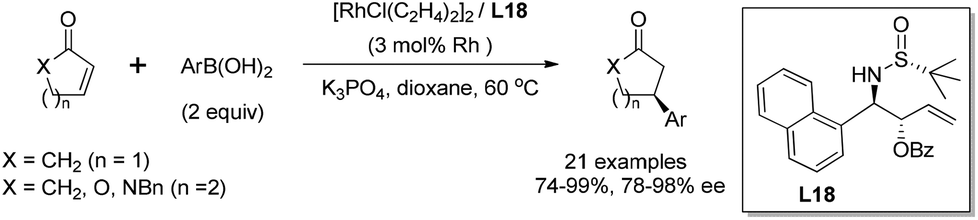 | ||
| Scheme 36 Asymmetric arylation of α,β-unsaturated cyclic carbonyl compounds catalyzed sulfinamide–olefin ligand. | ||
With the above significant success in the rhodium-catalyzed asymmetric 1,4-addition using chiral sulfur–olefin ligands, the exploration of this new ligand class and applying them in the rhodium-catalyzed enantioselective 1,2-addition of arylboronic acids to α-ketoesters and α-diketones was expanded.62 In the initial screening with methyl phenylglyoxylate and p-tolylboronic acid as the reaction substrates (Scheme 37), it was found that a simple sulfur–olefin ligand L21 could lead to 1,2-addition in high yield (90%) and with excellent enantioselectivity (85% ee). It is noteworthy that the chiral diene ligand (L1) and diphosphine ligand (BINAP) afforded the desired adduct with very low stereoselectivities (9% ee and 13% ee, respectively), in spite of excellent chemical yields (90% and 79%, respectively).
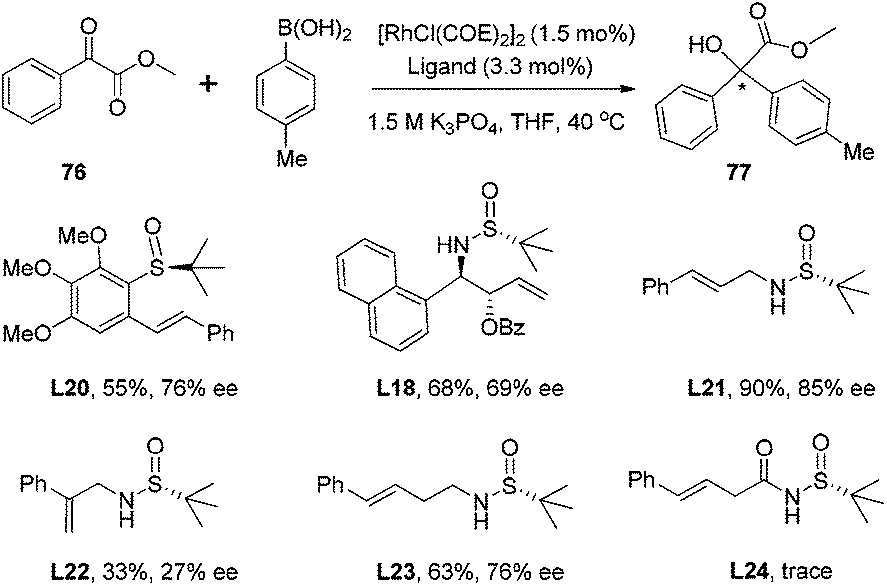 | ||
| Scheme 37 Ligand screening (COE = cyclooctene, Bz = benzoyl). | ||
The ester group in phenylglyoxylate showed some impact on the 1,2-addition in terms of chemical yield and enantioselectivity. Aromatic groups, such as phenyl, 4-chlorophenyl, 1-naphthyl and 2-naphthyl esters, afforded better enantioselectivities (92–95% ee), albeit with low yields (33–65%), due to basic hydrolysis when 0.5 equivalent of 1.5 M K3PO4 was applied at 40 °C. Further optimization of the base and reaction temperature led to the use of 2-naphthyl phenylglyoxylate as the best substrate, and the reaction could be accomplished at room temperature when 0.08 equivalent of 0.1 M KOH was applied, producing the desired tertiary α-hydroxyester in good yield (86%) and with excellent ee (94%).
With the identified optimal ester group and reaction conditions, the scope of different arylglyoxylates 78 and aryl boronic acids was investigated. As shown in Scheme 38, the 1,2-addition reactions consistently generated the corresponding adducts 79 in moderate to good yields (54–89%) and with excellent enantioselectivities (90–95% ee). It is noteworthy that both enantiomers could be obtained with high ee values by simply switching the corresponding Ar1 and Ar2 groups of the α-ketoesters and boronic acids.
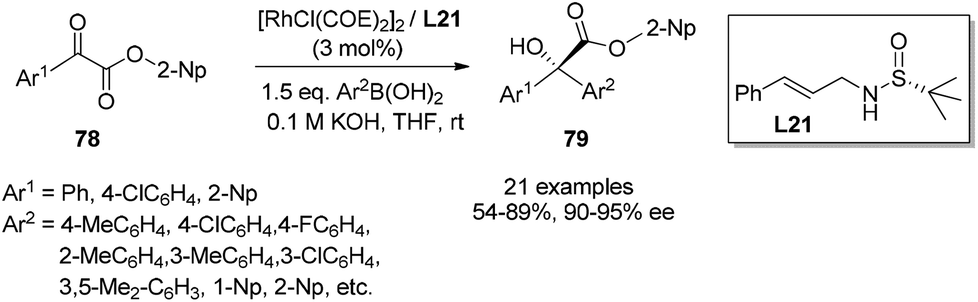 | ||
| Scheme 38 Catalytic asymmetric 1,2-addition of arylboric acids to α-ketoesters. | ||
This method could be extended to heteroaryl α-ketoesters.60b A variety of α-ketoesters including 3-indoleglyoxylates, 3-benzofuranglyoxylates and 3-benzothiopheneglyoxylates could react with arylboronic acids, affording corresponding quaternary carbon-containing heteroaromatic α-hydroxy esters in moderate to good yields with high enantiomeric excesses (up to 97%). As for the 1,2-addition to aliphatic α-ketoesters, moderate yields and enantioselectivities were also achieved despite the possible formation of enolate esters under basic reaction conditions.60c
This enantioselective 1,2-addition could also be applied to α-diketone substrates 80.60a As shown in Scheme 39, the enantioselectivities for di-aryl α-diketones were generally quite high (95–99% ee), while the less hindered aliphatic diketone, biacetyl (R1 = R2 = Me), gave the desired adduct with only 63% ee. For the asymmetric α-diketone (R1 = Me, R2 = Ph), the 1,2-addition predominantly took place at the less hindered carbonyl group (8.5:1 regioselectivity), affording the major adduct with 80% ee. Several bicyclic heterocycles with quaternary carbon-containing chiral center were successfully obtained through short or one-pot chemical transformations from such 1,2-additions of properly designed α-ketoesters and α-diketones.63
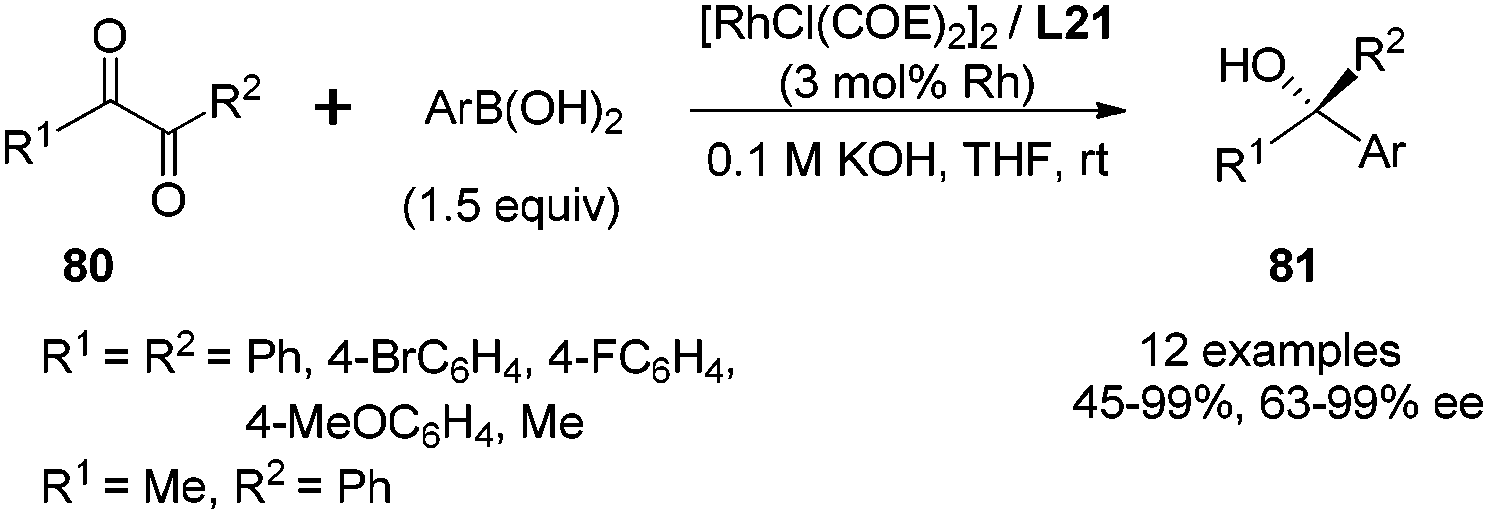 | ||
| Scheme 39 Catalytic asymmetric 1,2-addition of arylboric acids to α-diketones. | ||
Inspired by the success of enantioselective addition of arylboron reagents to CC and CO double bonds using simple chiral sulfur–olefin ligands, the attention was turned to their applications in the asymmetric 1,2-addition to CN double bonds to construct pharmacologically attractive chiral amines. Initially, we investigated the rhodium-catalyzed addition of arylboronic acids to cyclic N-sulfonyl α-iminoesters 82. Simple sulfur–olefins could successfully promote this arylation reaction. Under the optimized catalytic system, the reaction between a wide variety of cyclic N-sulfonyl α-iminoesters and arylboronic acids with diverse steric and electronic properties proceeded well, giving the corresponding addition products 83 mostly in high yields (up to 96%) and with excellent enantioselectivities (85–99% ee) (Scheme 40).64 Furthermore, the asymmetric arylation of a serial of other cyclic N-sulfonyl imines (84, 86, 88) were also subjected to this reaction. Although the reaction yields varied for different substrates, excellent enantioselectivities were observed in all examined cases. Notably, the benzo-fused six-membered cyclic imine 86 yielded particularly interesting CF3-containing cyclic sulfamidates.
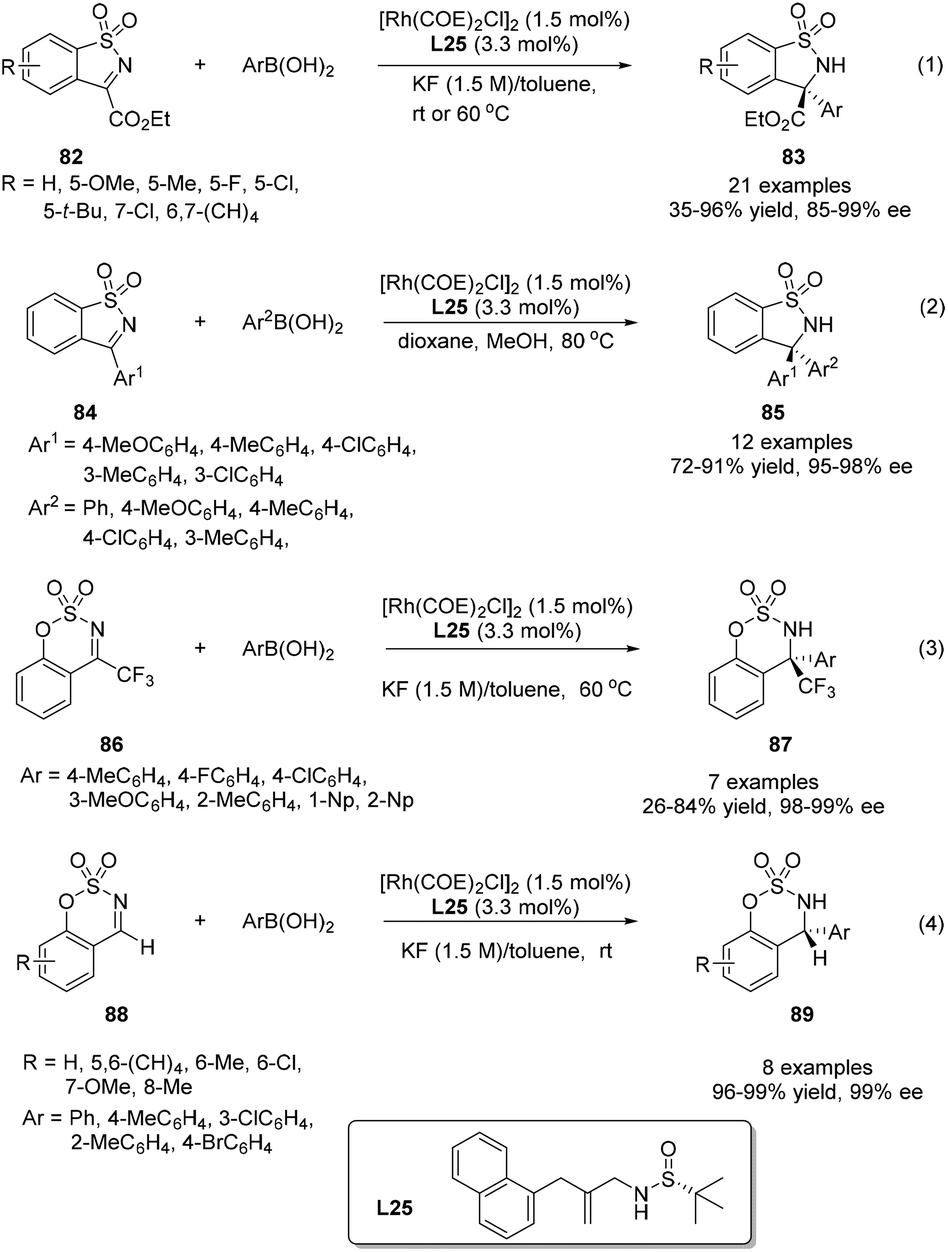 | ||
| Scheme 40 Catalytic asymmetric 1,2-addition of arylboric acids to cyclic N-sulfonyl imines. | ||
The application of chiral ligand L25 in the addition of arylboronic acids to 3,4-disubstituted thiadiazoles was also developed, providing direct access to chiral quaternary carbon-containing 1,2,5-thiadiazoline 1,1-dioxide derivatives (Scheme 41). The reactions proceeded under exceptionally mild conditions, giving the corresponding adducts in high yields and with excellent enantioselectivities.65
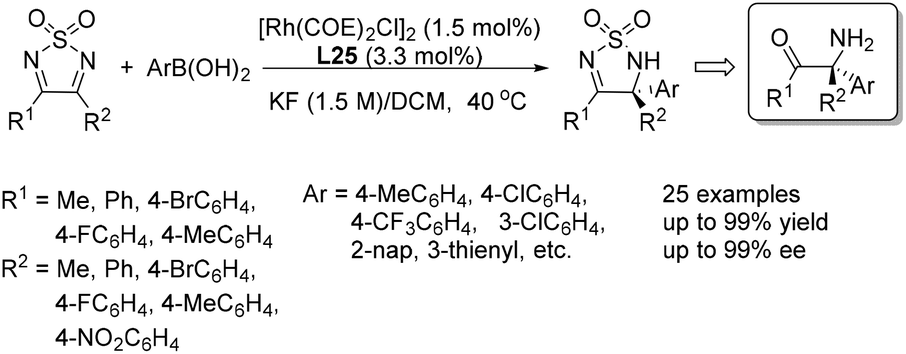 | ||
| Scheme 41 Asymmetric arylation of 3,4-disubstituted thiadiazoles catalyzed by sulfoxide–olefin ligand. | ||
Encouraged by the above results from chiral sulfinamide–olefin ligands, we designed and synthesized a new set of compounds with the sulfinyl group serving as the only chiral center to control the stereoselectivity.66 In this case, the chiral sulfinyl group and olefin were connected by a benzene ring in a 1,2-fashion (Fig. 7). These new ligands were evaluated in the rhodium-catalyzed 1,4-addition of phenylboronic acid to 2-cyclohexenone.
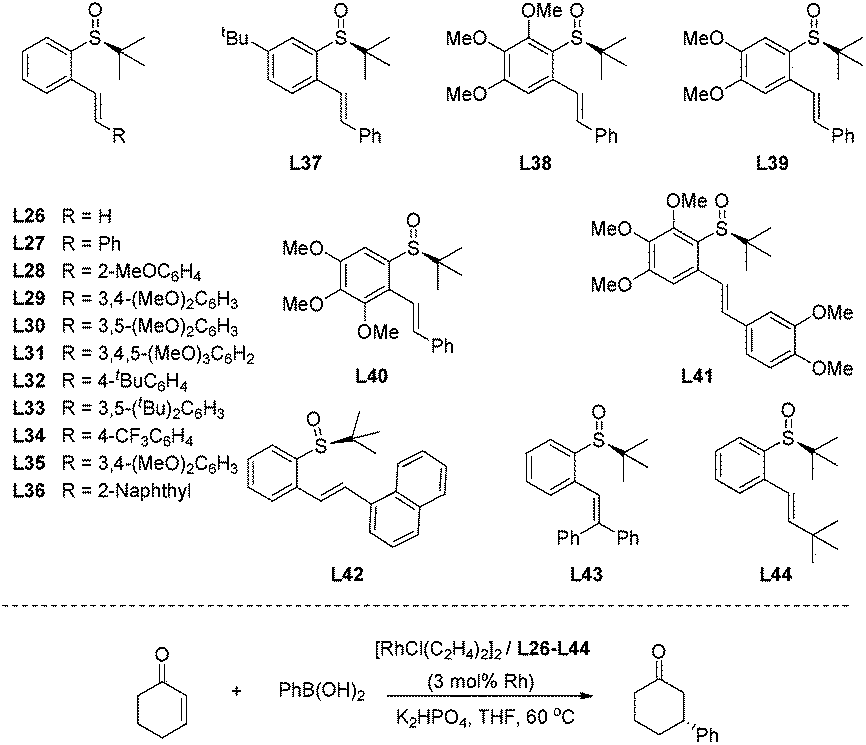 | ||
| Fig. 7 Sulfoxide–olefin ligands. | ||
The ligand with terminal olefin (L26, R = H) could promote the addition reaction in high yields (98%), albeit with quite low enantioselectivity (8% ee). To our delight, most of the ligands (L27–36) bearing R substituent displayed dramatic improvement in terms of enantioselectivities (70–95% ee). The bulky terminal group 1-naphthyl (L42) led to the desired product in only 4% yield and with 48% ee. The ligands with diphenyl (L43) and t-butyl groups (L44) at the olefin terminal only generated a negligible amount of product, suggesting that the bulky R groups would disfavor the coordination with rhodium. An evaluation of the different substitutions at the central benzene ring revealed that the introduction of multiple methoxy groups was beneficial for the reaction yield and enantioselectivity. The ligand L38, with a 2,3,4-trimethoxybenzene ring connecting the sulfinyl and olefin moieties, could afford the desired adduct in 99% yield and with the highest enantioselectivity (95% ee). Further optimization of the base and the reaction solvent for ligand L38 led to the enantioselectivity of 97% ee along with 99% yield when aqueous K2HPO4 and THF were used in this rhodium-catalyzed asymmetric arylation.
With the best ligand L38 and under the optimal reaction conditions, we examined the substrate scope of this rhodium-catalyzed asymmetric conjugate arylation (Scheme 42). The reactions performed well for α,β-unsaturated cyclic carbonyl compounds, such as 2-cyclohexenone, 2-cyclopentenone, α,β-unsaturated δ-lactone, giving the corresponding adducts with excellent enantioselectivities (94–98% ee), regardless of the electronic and steric properties of arylboronic acids. Unfortunately, the reaction of linear enone (E)-2-heptenone with phenyl boronic acid gave the desired product in a low yield (34%) and stereoselectivity (21% ee). The research results revealed that well-designed innovative S-stereogenic olefin ligands can be as useful as other classical chiral ligands in asymmetric transformations.
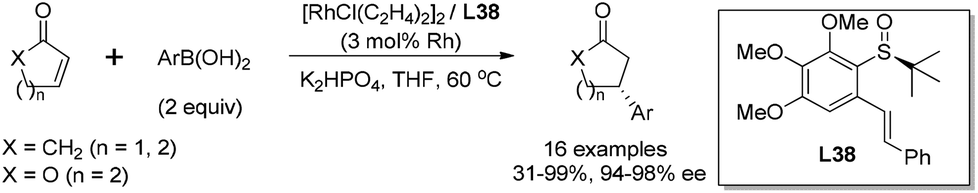 | ||
| Scheme 42 Asymmetric arylation of α,β-unsaturated cyclic carbonyl compounds catalyzed sulfoxide–olefin ligand. | ||
Inspired by the success of the above initial research on chiral sulfur–olefin ligands, further explorations aimed at expanding the applications of these new novel ligands have been carried out by other groups and us, which were described in detail in a recent review.4
5. Conclusion and outlook
The development of asymmetric synthesis with a highly enantioselective control continues to be one of the main topics in organic synthesis. Synthetic methods based on chiral auxiliaries provide reliable approaches to access chiral compounds. Using N-tert-butansulfinamide as a chiral auxiliary, we successfully developed three different kinds of stereoselective reactions to prepare enantiorich amines. SmI2-mediated homo-coupling and cross-coupling of N-tert-butansulfinyl imines with nitrones and aldehydes provide a simple and efficient way to synthesize enantiorich vicinal diamines and α-amino alcohols. As for the Zn/In-mediated allylation of N-tert-butansulfinyl imines, the stereoselectivity reversal can be controlled by switching the reaction conditions. More complicated chiral compounds can be synthesized by using suitable multifunctional allyl bromide reagents. Highly diastereoselective intermolecular or intramolecular aza-Friedel–Crafts reaction have also been realized in the presence of proper Lewis acid catalysts.In the meantime, highly enantioselective asymmetric transition-metal-catalyzed reactions were also pursued using structurally novel chiral dienes as ligands. Two families of chiral diene ligands bearing bicyclo[3.3.0]octadiene or dicyclopentadiene skeleton were successfully introduced. A variety of highly enantioselective rhodium-catalyzed reactions, including 1,2-addition to imines and 1,4-conjugate additions to several electron-deficient double bonds were performed. The first successful application of chiral diene ligands in palladium-catalyzed cross-coupling reactions was also realized.
Inspired by our success in these two parts, we developed conceptually new chiral sulfur–olefin ligands. These easily accessible new ligands proved to be competent for a variety of rhodium-catalyzed reactions, especially in the addition to specific CO and CN double bonds, allowing for highly enantioselective construction of otherwise-difficult-to-access fully-substituted carbon stereocenters.
Despite many successful examples of chiral N-tert-butanesulfinamide auxiliary and chiral diene and sulfur–olefin ligands, there are still many challenges we have to face. The application of both chiral auxiliary and ligands requires further expansion to cover more useful and challenging substrates. A more comprehensive mechanistic study on the catalytic role of ligands, as well as the development of new effective ligands, is still underway. Nevertheless, we believe that the power of these asymmetric reactions will be further demonstrated in organic synthesis and drug development.
Acknowledgements
Financial support from the National Natural Science Foundation of China (21361162001, 21325209), Shanghai Commission of Science and Technology (13JC1406900, 14XD1404400) and the State Key Laboratory of Bioorganic and Natural Products Chemistry is greatly acknowledged. We highly appreciate the scientific contributions from our co-workers, whose names mostly appear in the references.Notes and references
- (a) R. Noyori, Asymmetric Catalysis in Organic Synthesis, Wiley, New York, 2nd edn, 1994 Search PubMed; (b) E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Comprehensive Asymmetric Catalysis, Springer, Berlin, 1999, vols. I–III Search PubMed; (c) I. Ojima, Catalytic Asymmetric Synthesis, John Wiley & Sons, Hoboken/NJ, 3rd edn, 2010 Search PubMed.
- G.-Q. Lin, M.-H. Xu, Y.-W. Zhong and X.-W. Sun, Acc. Chem. Res., 2008, 41, 831 CrossRef CAS PubMed.
- C.-G. Feng, M.-H. Xu and G.-Q. Lin, Synlett, 2011, 1345 CAS.
- Y. Li and M.-H. Xu, Chem. Commun., 2014, 50, 3771 RSC.
- (a) J. A. Ellman, T. D. Owens and T. P. Tang, Acc. Chem. Res., 2002, 35, 984 CrossRef CAS PubMed; (b) J. A. Ellman, Pure Appl. Chem., 2003, 75, 39 CrossRef CAS; (c) C. H. Senanayake, D. Krishnamurthy, Z. H. Lu, Z. X. Han and I. Gallou, Aldrichimica Acta, 2005, 38, 93 CAS; (d) D. Morton and R. A. Stockman, Tetrahedron, 2006, 62, 8869 CrossRef CAS PubMed.
- (a) M.-H. Xu, W. Wang and G.-Q. Lin, Org. Lett., 2000, 2, 2229 CrossRef CAS PubMed; (b) W. Wang, M.-H. Xu, X.-S. Lei and G.-Q. Lin, Org. Lett., 2000, 2, 3773 CrossRef CAS PubMed; (c) M.-H. Xu, W. Wang, L.-J. Xia and G.-Q. Lin, J. Org. Chem., 2001, 66, 3953 CrossRef CAS PubMed.
- H. B. Kagan, Tetrahedron, 2003, 59, 10351 CrossRef CAS PubMed.
- Y. W. Zhong, K. Izumi, M. H. Xu and G. Q. Lin, Org. Lett., 2004, 6, 4747 CrossRef CAS PubMed.
- Y.-W. Zhong, M.-H. Xu and G.-Q. Lin, Org. Lett., 2004, 6, 3953 CrossRef CAS PubMed.
- Y.-W. Zhong, Y.-Z. Dong, K. Fang, K. Izumi, M.-H. Xu and G.-Q. Lin, J. Am. Chem. Soc., 2005, 127, 11956 CrossRef CAS PubMed.
- (a) Y.-W. Zhong, Y.-Z. Dong, K. Fang, K. Izumi, M.-H. Xu and G.-Q. Lin, J. Am. Chem. Soc., 2005, 127, 11956 CrossRef CAS PubMed; (b) R.-H. Liu, K. Fang, B. Wang, M.-H. Xu and G.-Q. Lin, J. Org. Chem., 2008, 73, 3307 CrossRef CAS PubMed; (c) R. Wang, K. Fang, B.-F. Sun, M.-H. Xu and G.-Q. Lin, Synlett, 2009, 2301 CAS.
- M. Lombardo and C. Trombini, Chem. Rev., 2007, 107, 3843 CrossRef CAS PubMed.
- (a) Amino Acids, Peptides, and Proteins in Organic Chemistry, ed. A. B. Hughes, Wiley-VCH, Weinheim, 2009, vol. 1 and 2 Search PubMed; (b) G. C. Barrett and D. T. Elmore, Amino Acids and Peptides, Cambridge University Press, Cambridge, 1998 Search PubMed; (c) F. P. J. T. Rutjes, L. B. Wolf and H. E. Schoemaker, J. Chem. Soc., Perkin Trans. 1, 2000, 4197 RSC.
- D.-M. Ji and M.-H. Xu, Chem. Commun., 2010, 46, 1550 RSC.
- Y. Li, D.-M. Ji and M.-H. Xu, Org. Biomol. Chem., 2011, 9, 8452 CAS.
- (a) S. Ni, Y. Yuan, J. Huang, X. Mao, M. Lv, J. Zhu, X. Shen, J. Pei, L. Lai, H. Jiang and J. Li, J. Med. Chem., 2009, 52, 5295 CrossRef CAS PubMed; (b) M. Sabbah, L. Soulre, S. Reverchon, Y. Queneau and A. Doutheau, Bioorg. Med. Chem., 2011, 19, 4868 CrossRef CAS PubMed; (c) X. Zhang, Y. He, S. Liu, Z. Yu, Z.-X. Jiang, Z. Yang, Y. Dong, S. C. Nabinger, L. Wu, A. M. Gunawan, L. Wang, R. J. Chan and Z.-Y. Zhang, J. Med. Chem., 2010, 53, 2482 CrossRef CAS PubMed.
- Y. Li and M.-H. Xu, Asian J. Org. Chem., 2013, 2, 50 CrossRef CAS.
- D. A. Cogan, G. C. Liu and J. Ellman, Tetrahedron, 1999, 55, 8883 CrossRef CAS.
- F. Foubelo and M. Yus, Tetrahedron: Asymmetry, 2004, 15, 3823 CrossRef CAS PubMed.
- X.-W. Sun, M.-H. Xu and G.-Q. Lin, Org. Lett., 2006, 8, 4979 CrossRef CAS PubMed.
- X.-W. Sun, M. Liu, M.-H. Xu and G.-Q. Lin, Org. Lett., 2008, 10, 1259 CrossRef CAS PubMed.
- M. Liu, X.-W. Sun, M.-H. Xu and G.-Q. Lin, Chem. – Eur. J., 2009, 15, 10217 CrossRef CAS PubMed.
- For reviews on the synthesis of β-amino alcohols: (a) D. J. Ager, I. Prakash and D. R. Schaad, Chem. Rev., 1996, 96, 835 CrossRef CAS PubMed; (b) C. H. Senanayake, Aldrichimica Acta, 1998, 31, 3 CAS; (c) S. C. Bergmeier, Tetrahedron, 2000, 56, 2561 CrossRef CAS; (d) G. Mlostoń, E. Obijalska and H. Heimgartner, J. Fluorine Chem., 2010, 131, 829 CrossRef PubMed.
- M. Liu, A. Shen, X.-W. Sun, F. Deng, M.-H. Xu and G.-Q. Lin, Chem. Commun., 2010, 46, 8460 RSC.
- W.-J. Liu, Y.-H. Zhao and X.-W. Sun, Tetrahedron Lett., 2013, 54, 3586 CrossRef CAS PubMed.
- (a) H. Kakeya, M. Morishita, H. Kobinata, M. Osono and H. Osada, J. Antibiot., 1998, 51, 1126 CrossRef CAS; (b) H. Kakeya, M. Morishita, H. Kobinata, T. Morita, K. Kobayashi and H. Osada, J. Org. Chem., 1999, 64, 1052 CrossRef CAS.
- For related study, see: L. R. Reddy, B. Hu, M. Prashad and K. Prasad, Org. Lett., 2008, 10, 3109 CrossRef CAS PubMed.
- (a) A. Shen, M. Liu, Z.-S. Jia, M.-H. Xu and G.-Q. Lin, Org. Lett., 2010, 12, 5154 CrossRef CAS PubMed; (b) R. Wang, P. Tian and G.-Q. Lin, Chin. J. Chem., 2013, 31, 40 CrossRef CAS; (c) Y. Tan, X.-D. Yang, W.-J. Liu and X.-W. Sun, Tetrahedron Lett., 2014, 55, 6105 CrossRef CAS PubMed.
- A. Shen, Z.-T. He, H.-J. Yu, Y. Fukui, P. Tian and G.-Q. Lin, Synlett, 2013, 1649 CAS.
- T. Guo, R. Song, B.-H. Yuan, X.-Y. Chen, X.-W. Sun and G.-Q. Lin, Chem. Commun., 2013, 49, 5402 RSC.
- D. Chen and M.-H. Xu, Chem. Commun., 2013, 49, 1327 RSC.
- Z.-Q. Wang, C.-G. Feng, M.-H. Xu and G.-Q. Lin, J. Am. Chem. Soc., 2007, 129, 5336 CrossRef CAS PubMed.
- S. Helbig, S. Sauer, N. Cramer, S. Laschat, A. Baro and W. Frey, Adv. Synth. Catal., 2007, 349, 2331 CrossRef CAS.
- C. Shao, H.-J. Yu, N.-Y. Wu, C.-G. Feng and G.-Q. Lin, Org. Lett., 2010, 12, 3820 CrossRef CAS PubMed.
- H.-Y. Yang and M.-H. Xu, Chin. J. Chem., 2013, 31, 119 CrossRef CAS.
- T. Hayashi, K. Ueyama, N. Tokunaga and K. Yoshida, J. Am. Chem. Soc., 2003, 125, 11508 CrossRef CAS PubMed.
- C. Fischer, C. Defieber, T. Suzuki and E. M. Carreira, J. Am. Chem. Soc., 2004, 126, 1628 CrossRef CAS PubMed.
- Reviews of chiral olefin ligands: (a) C. Defieber, H. Grutzmacher and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 4482 CrossRef CAS PubMed; (b) J. B. Johnson and T. Rovis, Angew. Chem., Int. Ed., 2008, 47, 840 CrossRef CAS PubMed; (c) R. Shintani and T. Hayashi, Aldrichimica Acta, 2009, 42, 31 CAS; (d) X. Q. Feng and H. F. Du, Asian J. Org. Chem., 2012, 1, 204 CrossRef CAS.
- Reviews on arylation of imines: (a) C. S. Marques and A. J. Burke, ChemCatChem, 2011, 3, 635 CrossRef CAS; (b) P. Tian, H.-Q. Dong and G.-Q. Lin, ACS Catal., 2012, 2, 95 CrossRef CAS.
- H.-Y. Yang and M.-H. Xu, Chem. Commun., 2010, 46, 9223 RSC.
- M. Trincado and J. A. Ellman, Angew. Chem., Int. Ed., 2008, 47, 5623 CrossRef CAS PubMed.
- Z. Cui, H.-J. Yu, R.-F. Yang, W.-Y. Gao, C.-G. Feng and G.-Q. Lin, J. Am. Chem. Soc., 2011, 133, 12394 CrossRef CAS PubMed.
- C. Shao, H.-J. Yu, N.-Y. Wu, C.-G. Feng and G.-Q. Lin, Org. Lett., 2010, 12, 3820 CrossRef CAS PubMed.
- Y. F. Luo, A. J. Carnell and H. W. Lam, Angew. Chem., Int. Ed., 2012, 51, 6762 CrossRef CAS PubMed.
- R. Shintani, M. Takeda, Y.-T. Soh, T. Ito and T. Hayashi, Org. Lett., 2011, 13, 2977 CrossRef CAS PubMed.
- Z. Cui, Y.-J. Chen, W.-Y. Gao, C.-G. Feng and G.-Q. Lin, Org. Lett., 2014, 16, 1016 CrossRef CAS PubMed.
- C.-G. Feng, Z.-Q. Wang, P. Tian, M.-H. Xu and G.-Q. Lin, Chem. – Asian J., 2008, 3, 1511 CrossRef CAS PubMed.
- C.-G. Feng, Z.-Q. Wang, C. Shao, M.-H. Xu and G.-Q. Lin, Org. Lett., 2008, 10, 4101 CrossRef CAS PubMed.
- C. Shao, H.-J. Yu, C.-G. Feng, R. Wang and G.-Q. Lin, Tetrahedron Lett., 2012, 53, 2733 CrossRef CAS PubMed.
- Z.-Q. Wang, C.-G. Feng, S.-S. Zhang, M.-H. Xu and G.-Q. Lin, Angew. Chem., Int. Ed., 2010, 49, 5780 CrossRef CAS PubMed.
- C. Shao, H.-J. Yu, N.-Y. Wu, P. Tian, R. Wang, C.-G. Feng and G.-Q. Lin, Org. Lett., 2011, 13, 788 CrossRef CAS PubMed.
- For rhodium-catalyzed asymmetric conjugate addition of alkenyl silicon and zirconium reagents, see: (a) S. Oi, Y. Honma and Y. Inoue, Org. Lett., 2002, 4, 667 CrossRef CAS PubMed; (b) S. Oi, A. Taira, Y. Honma and Y. Inoue, Org. Lett., 2003, 5, 97 CrossRef CAS PubMed; (c) Y. Otomaru and T. Hayashi, Tetrahedron: Asymmetry, 2004, 15, 2647 CrossRef CAS PubMed; (d) Y. Nakao, J. Chen, H. Imanaka, T. Hiyama, Y. Ichikawa, W.-L. Duan, R. Shintani and T. Hayashi, J. Am. Chem. Soc., 2007, 129, 9137 CrossRef CAS PubMed; (e) S. Oi, T. Sato and Y. Inoue, Tetrahedron Lett., 2004, 45, 5051 CrossRef CAS PubMed.
- For selected examples: (a) Y. Takaya, M. Ogasawara and T. Hayashi, Tetrahedron Lett., 1998, 39, 8479 CrossRef CAS; (b) A. Duursma, J. G. Boiteau, L. Lefort, J. A. F. Boogers, A. H. M. de Vries, J. G. de Vries, A. J. Minnaard and B. L. Feringa, J. Org. Chem., 2004, 69, 8045 CrossRef CAS PubMed; (c) R. T. Stemmler and C. Bolm, J. Org. Chem., 2005, 70, 9925 CrossRef CAS PubMed; (d) G. Lalic and E. J. Corey, Tetrahedron Lett., 2008, 49, 4894 CrossRef CAS PubMed; (e) K. Okamoto, T. Hayashi and V. H. Rawal, Org. Lett., 2008, 10, 4387 CrossRef CAS PubMed.
- H.-J. Yu, C. Shao, Z. Cui, C.-G. Feng and G.-Q. Lin, Chem. – Eur. J., 2012, 18, 13274 CrossRef CAS PubMed.
- S. Murakami, T. Takemoto and Z. Shimizu, J. Pharm. Soc. Jpn., 1953, 73, 1026 CAS.
- M. A. Grundl, J. J. Kennedy-Smith and D. Trauner, Organometallics, 2005, 24, 2831 CrossRef CAS.
- S.-S. Zhang, Z.-Q. Wang, M.-H. Xu and G.-Q. Lin, Org. Lett., 2010, 12, 5546 CrossRef CAS PubMed.
- X.-S. He, S.-S. Zhang, Y.-L. Guo, H.-Y. Wang and G.-Q. Lin, Organometallics, 2012, 31, 2945 CrossRef CAS.
- S.-S. Jin, H. Wang and M.-H. Xu, Chem. Commun., 2011, 47, 7230 RSC.
- (a) T. Thaler, L.-N. Guo, A. K. Steib, M. Raducan, K. Karaghiosoff, P. Mayer and P. Knochel, Org. Lett., 2011, 13, 3182 CrossRef CAS PubMed; (b) G.-H. Chen, J.-Y. Gui, L.-C. Li and J. Liao, Angew. Chem., Int. Ed., 2011, 50, 7681 CrossRef CAS PubMed; (c) F. Xue, X.-C. Li and B.-S. Wan, J. Org. Chem., 2011, 76, 7256 CrossRef CAS PubMed; (d) X. Q. Feng, Y. Z. Wang, B. B. Wei, J. Yang and H. F. Du, Org. Lett., 2011, 13, 3300 CrossRef CAS PubMed.
- S.-S. Jin, H. Wang, T.-S. Zhu and M.-H. Xu, Org. Biomol. Chem., 2012, 10, 1764 CAS.
- (a) T.-S. Zhu, S.-S. Jin and M.-H. Xu, Angew. Chem., Int. Ed., 2012, 51, 780 CrossRef CAS PubMed; (b) H. Wang, T.-S. Zhu and M.-H. Xu, Org. Biomol. Chem., 2012, 10, 9158 RSC; (c) T.-S. Zhu and M.-H. Xu, Chin. J. Chem., 2013, 31, 321 CrossRef CAS.
- (a) Y. Li, D.-X. Zhu and M.-H. Xu, Chem. Commun., 2013, 49, 11659 RSC; (b) T.-S. Zhu, J.-P. Chen and M.-H. Xu, Chem. – Eur. J., 2013, 19, 865 CrossRef CAS PubMed.
- (a) H. Wang, T. Jiang and M.-H. Xu, J. Am. Chem. Soc., 2013, 135, 971 CrossRef CAS PubMed; (b) H. Wang and M.-H. Xu, Synthesis, 2013, 2125 CAS.
- H. Wang, Y. Li and M.-H. Xu, Org. Lett., 2014, 16, 3962 CrossRef CAS PubMed.
- W.-Y. Qi, T.-S. Zhu and M.-H. Xu, Org. Lett., 2011, 13, 410 CrossRef PubMed.
Footnote |
| † Present address: Arvinas Inc., 5 Science Park, New Haven, CT 06511, USA. |
| This journal is © the Partner Organisations 2015 |