
Magnetic ordering in TCNQ-based metal–organic frameworks with host–guest interactions†
Xuan
Zhang
 a,
Mohamed R.
Saber
a,
Andrey P.
Prosvirin
a,
Joseph H.
Reibenspies
a,
Lei
Sun
b,
Maria
Ballesteros-Rivas
a,
Hanhua
Zhao
a and
Kim R.
Dunbar
*a
a,
Mohamed R.
Saber
a,
Andrey P.
Prosvirin
a,
Joseph H.
Reibenspies
a,
Lei
Sun
b,
Maria
Ballesteros-Rivas
a,
Hanhua
Zhao
a and
Kim R.
Dunbar
*a
aDepartment of Chemistry, Texas A&M University, College Station, TX 77842-3012, USA. E-mail: dunbar@chem.tamu.edu
bDepartment of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA
First published on 3rd September 2015
Abstract
Host–guest interactions between the aromatic molecules benzene, toluene, aniline and nitrobenzene and the redox-active TCNQ-based metal–organic framework (MOF), Fe(TCNQ)(4,4′-bpy) (1) (TCNQ = 7,7,8,8-tetracyanoquinodimethane), have been found to modulate spontaneous magnetization behaviours at low temperatures. An analogous MOF, Mn(TCNQ)(4,4′-bpy) (2) with isotropic Mn(II) ions as well as the two-dimensional compound Fe(TCNQ)(DMF)2·2DMF (3·2DMF), were also prepared as models for studying the effects of single-ion magnetic anisotropy and structural distortion on spin canting. The results indicate guest-dependent long range magnetic ordering occurs at low temperatures, which correlates with the electrostatic and steric effects of the incorporated aromatic guests.
Introduction
Metal–organic frameworks (MOFs), also known as porous coordination polymers (PCPs), have attracted enormous attention over the past two decades because of their potential applications in gas separation/storage, catalysis and sensing.1 These versatile crystalline materials exhibit rich structural variability and porosity and are ideal platforms for introducing multi-functionality. The topic of MOF materials has primarily focused on increasing porosity for gas storage but recent exciting advances are being made in relatively underexplored areas of catalytic reactivity,2 electrical conductivity3 and magnetic properties.4 Ligand design is extremely important in MOF research due to the plethora of choices these organic linkers offer for the variation of MOF topologies and functionalities. Long rigid anionic carboxylate and phosphonate, and/or neutral N-donor ligands have been commonly used as MOF linkers in order to ensure both large void spaces and thermodynamic stability of the frameworks.Recently, the use of redox-active organic linkers or those equipped with functional groups has been on the upsurge as part of the current trend to design MOF's that display special properties such as selective gas/guest adsorption, catalytic reactivity, charge mobility and conductivity.5 The use of redox-active linkers as alternatives to carboxylic acid ligands in MOFs is exemplified by the implementation of ligands such as TTF derivatives, NDIs, TCNQ˙−, TCNQ2− and (TCNQ-TCNQ)2− (TTF = tetrathiafulvalene, NDI = naphthalenediimide, TCNQ = 7,7,8,8-tetracyanoquino-dimethane) in the formation of MOFs.3a,6
The Investigation of MOF materials for different applications in magnetism, such as data storage and quantum computing,7 is hindered by the fact that the long linkers typically employed in the syntheses of MOF materials are not effective at mediating magnetic interactions. To tackle this problem, we have been studying organocyanide linkers that are known to engender conducting and magnetic properties in extended architectures and which have potential applications in non-volatile switching and memory devices.6c,8 The tunability of the redox activity of the organocyanide ligands make them very promising for accessing materials that combine semiconducting properties and magnetic ordering. Both theoretical and experimental studies have demonstrated that doubly reduced diamagnetic organocyanide ligands are capable of engendering effective magnetic coupling between metal spins, results that are attributed to an energetically favorable match between the ligand pπ and the metals dπ orbitals.9 In this vein, we and another group recently studied the magnetic properties of a series of anionic frameworks of the type [M2(TCNQ)3]2− (M = Mn, Fe, Co, Ni) and it was found that the TCNQ2− ligand promotes long range magnetic ordering despite the long coupling pathway.10 The lack of accessible cavities in these three-dimensional materials, however, precluded the possibility of enhancing magnetic and electronic properties via post-synthetic modifications or interactions with small guest molecules.
With the backdrop of previous work as a source of inspiration, we turned our attention to guest responsive magnetic MOFs as interesting targets because of their potential applications in molecular recognition and sensing in addition to the possibility for fine-tuning the magnetic properties by absorption/release of small guest molecules.11 Previously, we had found that [Mn2(TCNQF4)(CH3OH)7.5(H2O)0.5](TCNQF4)2·7.5CH3OH acts as a “magnetic sponge” and can reversibly switch between a glassy magnetic and a paramagnetic state triggered by de-solvation and solvation effects.12 Of interest in the context of this work is the existence of TCNQ dianionic-based MOFs, M(TCNQ)(4,4′-bpy) ⊃ CH3OH (M = Zn, Cd, Mn, Fe, Co; 4,4′-bpy = 4,4′-bipyridine),6g,13 which exhibit 3D structures composed of 2D MII-TCNQ2− neutral networks pillared by 4,4′-bpy (4,4′-bipyridine) linkers. It has been shown that these redox-active MOF materials preferentially absorb aromatic solvent molecules such as benzene, nitrobenzene and anisole, leading to distinct color changes owing to variable degrees of charge-transfer between the host frameworks and the guest molecules.13a We postulated that host–guest charge-transfer interactions could lead to guest-induced magnetic responses of the M-TCNQ grids of the MOFs based on changes in the electron density on the TCNQ dianion.
Herein, we report the guest modulated magnetic properties of the MOFs, M(TCNQ)(4,4′-bpy) ⊃ solvent (M = Fe (1), Mn(2)), with the inclusion of different aromatic solvent molecules as well as a new 2-D network compound, Fe(TCNQ)(DMF)2·2DMF (3, DMF = N,N-dimethylformamide). The TCNQ2− anions were found to promote cooperative interactions between the high-spin FeII ions modulated by the aromatic guest molecules. This case study of solvent mediated magnetic behavior of redox-active MOFs resulting from electrostatic and steric interactions between the framework and the guest molecules serves as a proof of concept for the realization of guest controlled magnetic MOFs.
Results and discussion
Syntheses and structures
The formation of TCNQ2− containing coordination networks has been achieved by disproportionation of TCNQ˙−, the deprotonation of H2TCNQ by lithium acetate and the direct reduction of neutral TCNQ by metal complexes.9b,13d,e The synthetic method employed in this work is slightly modified from reported procedures and was carried out using 4,4′-bpy/DMF in the absence of LiOAc to deprotonate H2TCNQ in order to generate TCNQ2−in situ. Under anaerobic conditions, layering of a solution of FeSO4·7H2O in CH3OH onto a mixture of H2TCNQ and 4,4′-bpy in DMF results in the formation of red-orange crystals of the product FeII(TCNQ)(4,4′-bpy) ⊃ CH3OH (1 ⊃ CH3OH) in three days. The reaction is considerably slower than the reported procedures with either LiOAc as the deprotonation reagent for H2TCNQ or the disproportionation reactions TCNQ radicals to produce TCNQ2−, a favorable situation which leads to the formation of larger single crystals.The structure of the compound (1 ⊃ CH3OH) was confirmed to be the same as previously reported by Kitagawa and coworkers.13d Data collected at 133 K reveal that the (FeII-TCNQ2−) units form corrugated sheets with adjacent μ4-TCNQ2− bridges being arranged perpendicular to each other (Fig. 1a); these sheets become more flattened as the temperature increases from 133 to 243 K. The 2-D networks are pillared by 4,4′-bipyridine to form the 3-D frameworks (Fig. 2) and the cavities are filled with methanol molecules that can be replaced with aromatic guest molecules by soaking the crystals in neat aromatic solvents. Single crystals are preserved during these experiments but only in the case of Zn(TCNQ)(4,4′-bpy) ⊃ C6H6 were the benzene molecules crystallographically located.13a In the case of the reported nitrobenzene and anisole soaked crystals of compound 1, the framework structure was solved with the Fe-TCNQ network being disordered between the corrugated sheets and the planar sheets; weak peaks of electron density were observed in the cavity and are ascribed to disordered guest molecules. Charge-transfer between the TCNQ2− in the host framework and the guest aromatic molecules was reported previously on the basis of diffuse reflectance UV-Vis spectra but no additional properties were reported.13d
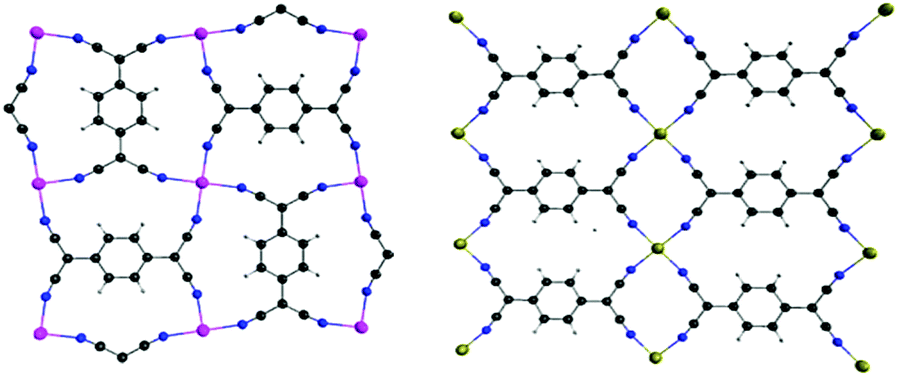 | ||
| Fig. 1 Two of the most common coordination modes of TCNQ dianions, with adjacent TCNQ species perpendicular (left) and parallel (right) to each other. | ||
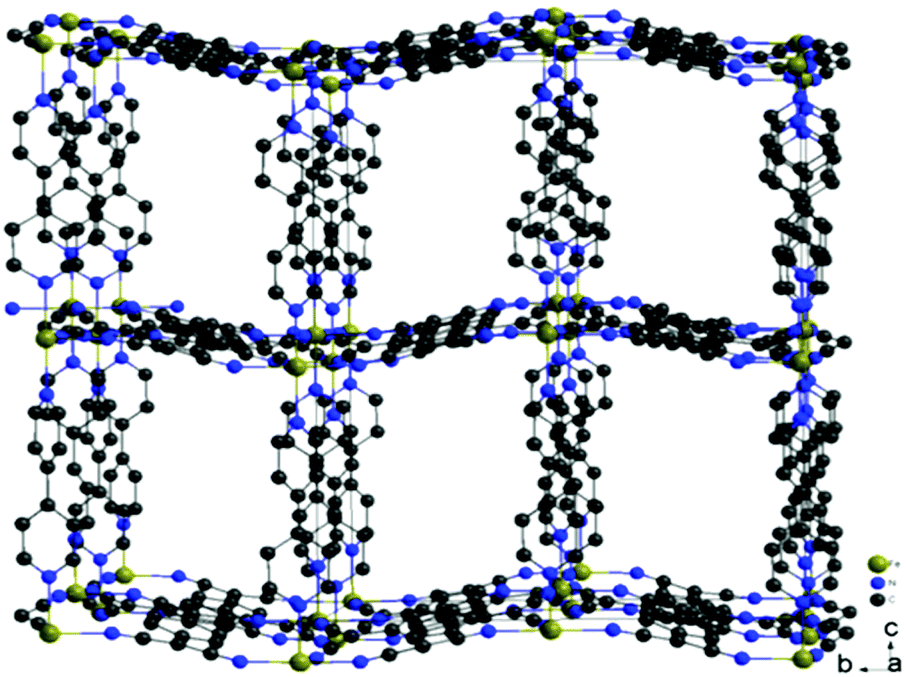 | ||
| Fig. 2 Structure of the metal–organic frameworks Fe(TCNQ)(4,4′-bpy) ⊃ CH3OH. Solvent molecules are omitted for clarity. | ||
We collected single crystal X-ray structures of compound 1 with different interstitial aromatic solvents in order to probe structure–property relationships (Table 1). For benzene, toluene and aniline, low electron densities of the disordered solvent molecules were observed in the frameworks. The structures were solved in the tetragonal space group I4/mcm, which is consistent with reported structures of these frameworks.13d Refinement of the guest molecules in the pores of the framework was carried out by using models of the corresponding aromatic molecules to fit the residual electron densities. The positioning of the guest molecules from refinement of 2 ⊃ C6H6 is consistent with the structure of Zn(TCNQ)(4,4′-bpy) ⊃ C6H6 as previously reported (Fig. 3, right), where no intimate interaction between the aromatic guest molecules and the host framework is observed and the planes of the aromatic molecule are vertical to the M-TCNQ 2-D planes. Unfortunately, the crystal structure of compound 1 ⊃ C6H5NO2 exhibited severe twinning and disorder problems and was not refined satisfactorily.† A possible lower symmetry space group was found, as compared to benzene, toluene and aniline containing frameworks of 1, which is consistent with the observation of extra peaks for 1 ⊃ C6H5NO2 in the powder X-ray diffraction patterns (vide infra).
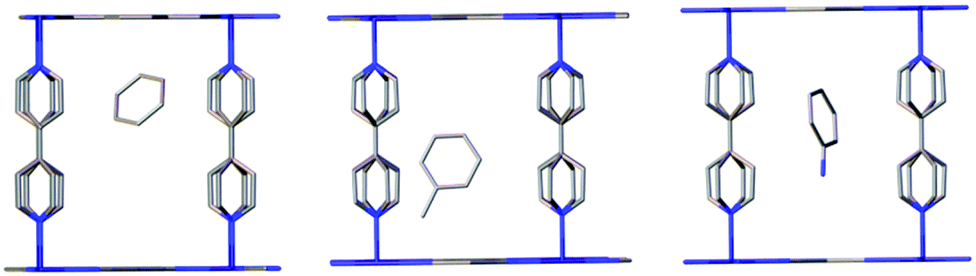 | ||
| Fig. 3 Fragments of the crystal structure of 1 ⊃ guest (benzene (left), toluene (middle) and aniline (right)) showing the relative positions of the different guest molecules in the MOF. The pyridyl and benzene rings are disordered, and only one of the disordered guest molecules are shown for the sake of clarity. | ||
| Compound | 1 ⊃ benzene | 1 ⊃ aniline | 1 ⊃ toluene |
|---|---|---|---|
| Empirical formula | C42FeN6H32 | C34H26FeN8 | C36H28FeN6 |
| Formula weight | 676.58 | 602.48 | 600.49 |
| Temperature/K | 100 | 110 | 100 |
| Crystal system | Tetragonal | Tetragonal | Tetragonal |
| Space group | I4/mcm | I4/mcm | I4/mcm |
| a/Å | 12.3580(6) | 12.340(2) | 12.3673(3) |
| c/Å | 22.9288(15) | 22.852(4) | 22.9031(6) |
| Volume/Å3 | 3501.7(4) | 3480.0(13) | 3503.03(19) |
| Z | 4 | 4 | 4 |
| Goodness-of-fit on F2 | 1.182 | 1.335 | 1.122 |
| Final R indexes [I ≥ 2σ (I)] | R 1 = 0.0598, wR2 = 0.1639 | R 1 = 0.0774, wR2 = 0.2042 | R 1 = 0.0592, wR2 = 0.1632 |
In addition to the relative positions of the guest molecules in the framework, structural distortions were also observed with the inclusion of different guest molecules as reflected in the bond distances and angles listed in Table S2.† The Fe-TCNQ 2-D sheets of the benzene, toluene and aniline containing MOFs are essentially planar with respect to the TCNQ2− bridges, whereas those of 1 ⊃ CH3OH and 1 ⊃ C6H5NO2 are clearly corrugated with larger TCNQ dihedral angles. Presumably the observed structural distortions reflect both steric and electrostatic effects of the guest molecules and are expected to exert different effects on the magnetic coupling through the TCNQ2− bridges as well as the canting angles of the Fe(II) centers.
In order to further investigate the effects of structural distortions as well as the single-ion magnetic anisotropy of the metal center, two additional model compounds were prepared, viz., Mn(TCNQ)(4,4′-bpy) (2)13e with the isotropic MnII ions and the new 2D compound Fe(TCNQ)(DMF)2·2DMF (3·2DMF) with planar Fe-TCNQ networks. As in the case of the FeII analogues, crystallographic studies of 2 ⊃ CH3OH revealed corrugated 2-D Mn-TCNQ networks whereas a new structure for 2 ⊃ C6H6 was found to contain planar Mn-TCNQ networks with disordered benzene molecules situated vertical to the Mn-TCNQ planes (Fig. S11,† left). These results further support the electrostatic nature of the interactions. Single crystal X-ray studies of compound 3·2DMF revealed a 2D structure with flat Fe-TCNQ sheets (the TCNQ2− units are parallel to each other as shown in Fig. 1b, Fe–N(cyano) bond lengths are 2.124(5) and 2.113(6) Å), with two DMF molecules occupying the axial positions (Fig. 4).
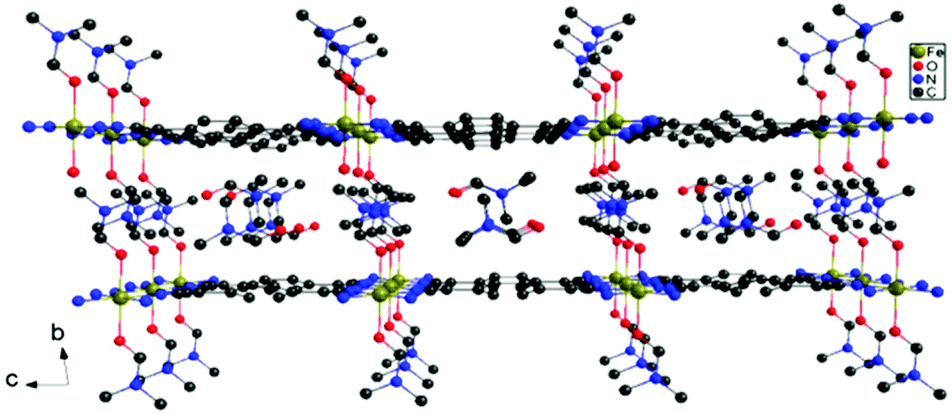 | ||
| Fig. 4 Packing diagram of 3 showing the flat planes of FeII-TCNQ2−. | ||
Powder X-ray diffraction
Room temperature powder X-ray diffraction methods were employed to monitor the structures of bulk samples containing different aromatic solvent molecules. The match of the patterns indicates that the frameworks of the structures are preserved in all cases (Fig. 5). Several new peaks arising from the inclusion of different solvent molecules may be attributed to the ordered arrangement of the corresponding solvent molecules and the small distortions of the host frameworks resulted from the included guest molecules.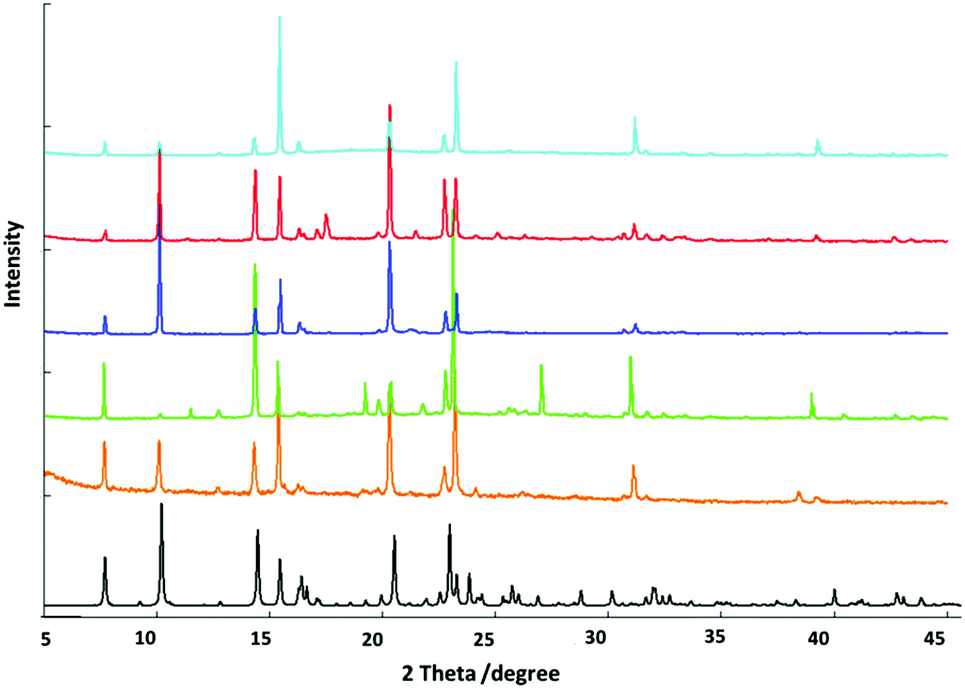 | ||
| Fig. 5 Powder X-ray diffraction patterns of Fe(TCNQ)(4,4′-bpy) with the inclusion of different solvent molecules: methanol (black, simulated), methanol (orange, experimental), benzene (blue), toluene (red), nitrobenzene (green) and aniline (turquoise). | ||
Infrared spectroscopy
Infrared spectral data, summarized in Table 2, indicate the presence of ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N) stretching modes characteristic of TCNQ2−.10 The lower energies of these bands as compared to previously reported compounds indicate a slightly higher negative charge on the TCNQ2− units, consistent with charge-transfer from aromatic guest molecules to the TCNQ2− units.
N) stretching modes characteristic of TCNQ2−.10 The lower energies of these bands as compared to previously reported compounds indicate a slightly higher negative charge on the TCNQ2− units, consistent with charge-transfer from aromatic guest molecules to the TCNQ2− units.
| Compound | ν(CN)/cm−1 |
|---|---|
| H2TCNQ | 2257, 2204 |
| 1 ⊃ CH3OH | 2181, 2112 |
| 1 ⊃ C6H6 | 2185, 2059 |
| 1 ⊃ C6H5NO2 | 2183, 2114 |
| 1 ⊃ C7H8 | 2183, 2116, 2055 |
| 1 ⊃ C6H5NH2 | 2182, 2116 |
| 3 | 2197, 2134 |
Magnetic properties
The tendency of the compounds in this study to lose interstitial solvents renders it difficult to accurately characterize them in the dry state. Owing to this situation, samples for magnetic measurements were conducted on crushed crystals covered in a minimum volume of the solvent in sealed quartz tubes to avoid interstitial solvent loss. The magnetic measurements of these compounds were carried out using a SQUID MPMS instrument over the temperature range 1.8–300 K. Diamagnetic contributions of the intercalated and excess solvent were accounted for during the subsequent data analysis process.The temperature dependence of the χT susceptibilities are shown in Fig. 6. The room temperature χT values (1 ⊃ CH3OH, 3.32 emu K mol−1; 1 ⊃ C6H6, 3.41 emu K mol−1; 1 ⊃ C7H8, 3.33 emu K mol−1; 1 ⊃ C6H5NO2, 3.48 emu K mol−1; 1 ⊃ C6H5NH2, 3.41 emu K mol−1) correspond to the expected value for an isolated high spin iron(II) center (S = 2, g = 2.12). Upon lowering the temperature, the χT values decrease monotonically down to ∼5 K (Fig. 6) indicative of antiferromagnetic interactions. The small fluctuations in the χT values above 50 K are attributed to freezing of the solvents. The temperature dependence of the magnetic susceptibilities between 300–5 K were fit to a Curie–Weiss law with Weiss constant values of θ = −9.4 K for 1 ⊃ CH3OH, −11.1 K for 1 ⊃ C6H5NO2 and −11.2 K for all three of 1 ⊃ C6H6, 1 ⊃ C7H8 and 1 ⊃ C6H5NH2 (Table 3), data that support the presence of antiferromagnetic interactions within the MOF framework (Table 3), in addition to other possible contributing factors such as spin–orbit coupling and also zero-field splitting at low temperatures.
 | ||
| Fig. 6 Temperature dependence of the χT plot of the MOF Fe(TCNQ)(4,4′-bpy) in different solvents. | ||
| Guest | T c/K | θ/K | J/cm−1 (estimated) | J/cm−1 (calculated) | Canting angle/° |
|---|---|---|---|---|---|
| Methanol | 4.5(2) | −9.4 | −0.41 | −0.32 | 0.02 |
| Aniline | 4.9(5) | −11.2 | −0.49 | −0.32 | 0.19 |
| Benzene | 3.9(2) | −11.2 | −0.49 | −0.35 | 0.42 |
| Toluene | 3.7(2) | −11.2 | −0.49 | −0.37 | 0.49 |
| Nitrobenzene | 4.7(2) | −11.1 | −0.48 | −0.34 | 0.84 |
For estimation of the magnetic exchange parameter the mean field approximation formula14 can be used:
| J = 3θkB/(2zS(S + 1)). | (1) |
Given the crystal structure, the magnetic susceptibility data can be alternatively interpreted through eqn (2), describing the χT of a 2D Heisenberg quadratic-layer antiferromagnet. This formula, based on the isotropic Heisenberg Hamiltonian H = −2JΣijSiSj is as follows:15
| Ng2β2/2χT = 3x+∑Cn/xn−1 | (2) |
Similar behavior was observed in our recently reported [Ph3PMe]2[Fe2(TCNQ)3] magnetic MOF,10 but it is worth noting that the estimated coupling constants using the same equation for the current series is more than double of that in the former case. This is presumably due to the higher degree of planarity of the bridging TCNQ units, which allows for better dπ–pπ overlap between the metal centers and TCNQ dianions. In the case of [Ph3PMe]2[Fe2(TCNQ)3] the planarity of the TCNQ2− moiety has been significantly disrupted (the dicyanomethyl and the phenyl fragments of TCNQ2− have an average dihedral angle of 28.05°), whereas in compound 1, the TCNQ2− moiety remains planar with only a little distortion in the case of methanol and nitrobenzene (see dihedral angles in Table S2†). Therefore, two additional magnetic superexchange coupling pathways (through trans-7,8- and cis-7,8-cyano groups) may be contributing to the dominating antiferromagnetic interactions in the temperature range of 300–5 K in addition to the 7,7-cyano coupling pathway through the short end of TCNQ2− as found in the case of [Ph3PMe]2[Fe2(TCNQ)3]. The additional coupling pathways can help to explain the increased coupling in the current series. Given the structural similarity of the dicyanomethyl fragment of the TCNQ2− bridge and dicyanamide anion, we note that they exhibit comparable magnetic coupling strength between Fe(II) spin centers.16
Below 5 K, maxima of χT begin to appear for all samples indicating the onset of weak ferromagnetic interactions. Taking into account that the magnetic interactions of the metal ions through 4,4′-bpy are usually weakly antiferromagnetic,8d,17 the magnetic behavior is essentially 2-D between the FeII centers in the frameworks bridged by μ4-TCNQ2− linkers; the increase in χT at low temperatures can be ascribed to canted spin states from the interlayer interactions. Structural distortions in the 2-D sheets of Fe(TCNQ) and/or electronic interactions between TCNQ2− and the aromatic solvent molecules are the plausible reasons for the uncompensated magnetic moments and therefore “weak ferromagnetic” (canted antiferromagnetic) responses at 3–5 K.
Zero-field-cooled (ZFC) and field-cooled (FC) magnetization data at 10 Oe exhibit bifurcations in all cases (Fig. S1†), implying that long-range ordering of the magnetic moments is occurring. This conclusion is also supported by the presence of frequency independent peaks in both the in-phase and out-of-phase AC magnetic susceptibilities at low temperatures which suggests a magnetic transition is occurring from a paramagnetic state with antiferromagnetic interactions to a canted antiferromagnetic state (Fig. 7). In addition, field dependent magnetization hysteresis loops were observed for all samples at 1.8 K (Fig. S3†), which signifies the presence of spontaneous magnetization below their corresponding Tc values.
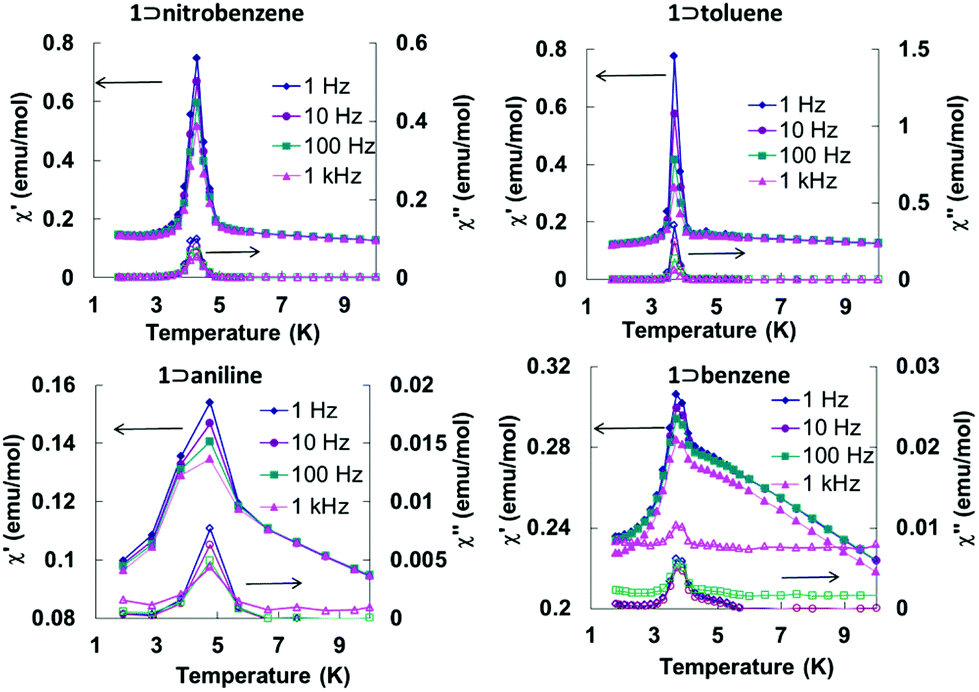 | ||
| Fig. 7 Variable temperature in-phase and out-of-phase AC magnetic susceptibility data for the Fe(TCNQ)(4,4′-bpy) MOFs in four aromatic solvents at different AC frequencies. | ||
The field dependent magnetization curves at 1.8 K (Fig. S4†) did not reach saturation even under a field of 7 T which is another piece of evidence for uncompensated moments from the spin-canted states. The canting angles (Table S2†) can be estimated from the equation ψ = tan−1(Mr/Ms), where Ms = gS is the saturation magnetization when all the moments are aligned in a parallel manner in the structure and Mr is the remnant magnetization (intercept of the linear part of magnetization curve).18 The largest canting angles of the 1 ⊃ nitrobenzene and 1 ⊃ toluene are consistent with the most prominent weak ferromagnetic responses at low temperatures.
The trend of ordering temperatures in the present series is affected by structural distortions of the Fe(TCNQ) planes which are the main source of spin canting. The effect of structural distortions is evident in 1 ⊃ CH3OH and 1 ⊃ C6H5NO2 which exhibit corrugated Fe(TCNQ) sheets unlike the planar sheets in the structures containing benzene, toluene and aniline. It is worth noting that the three polar solvents, CH3OH, C6H5NO2 and C6H5NH2 induce higher Tc values (4.5, 4.7 and 4.9 K respectively) as compared to the less polar benzene and toluene guest molecules (Tc = 3.9 and 3.7 K, respectively). Such an observation suggests an effect of guest polarity (Table S3†) on the ordering temperature which is consistent with the orientation of the guest molecules observed in the crystal structures. Another possible factor is charge-transfer between the host frameworks and the guest molecules which was previously reported.13d Such interactions should affect the spin density on the TCNQ2− units hence the magnetic coupling through it, but the observed Tc values do not simply follow the order of π-accepting strength of the guest molecules. It is also possible that the electrostatic interaction between the framework and guest molecules results in changes of the electron density on the TCNQ2−, and in turn affects magnetic coupling.
The relationship between the distortion of the structures and the magnetic properties can be further modeled for the 2-D network compound 3 which exhibits flat instead of corrugated planes of Fe-TCNQ units (Fig. 4). The hypothesis that structural distortion leads to the spin-canted states in the magnetically coupled 2-D network is further supported by this model compound. Temperature dependent magnetic measurements performed on 3 showed it to be a paramagnet with antiferromagnetic interactions with no spin-canted states at low temperatures. However, the χT value of a dry sample (3a) exhibits an increase at low temperatures (Fig. 8a), indicating the onset of a spin-canted antiferromagnetic state, which is probably due to the distortion of the flat plane to a corrugated plane upon the loss of DMF molecules. The lack of saturation of the magnetization for both samples indicates that the net magnetic moments stem from uncompensated canted spin states. The observation of a hysteresis loop and bifurcation in ZFC-FC curve (Fig. 8b and S2†) for the dry sample 3a also underscores the importance of structural distortion for the canted spin states in the present series.
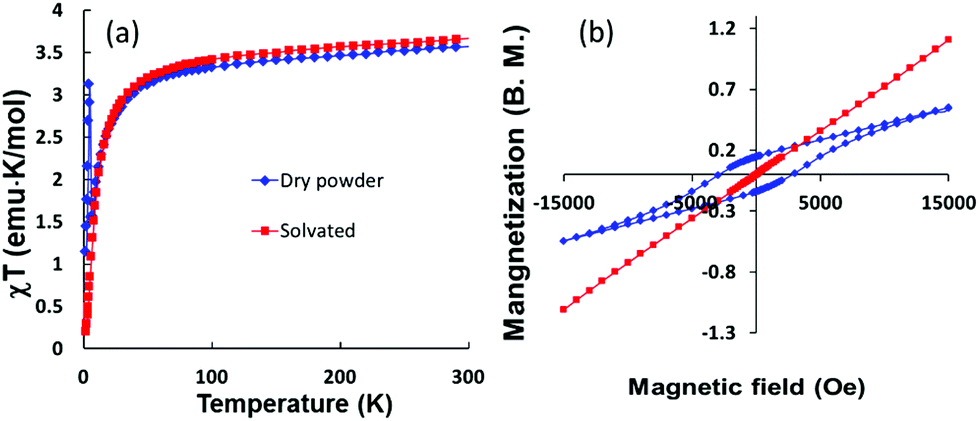 | ||
| Fig. 8 Temperature dependence of the χT product (left) nd hysteresis of the magnetization (right) of Fe(TCNQ)(DMF)2·2DMF. | ||
Single-ion magnetic anisotropy was also found to play a role in the canted states of the Fe(TCNQ)(bpy) MOFs. When the isotropic MnII analog was used, 2 ⊃ CH3OH showed only antiferromagnetic ordering at low temperatures without the presence of the canted spin states although the 2-D networks of Mn-TCNQ are considerably corrugated.
Experimental
The synthetic procedures were carried out under a nitrogen atmosphere using standard Schlenk-line techniques. H2TCNQ was prepared according to the literature by the reduction of TCNQ with mercaptoacetic acid.19 Fe(TCNQ)(4,4′-bpy)·6CH3OH was prepared by a slight modification of the reported procedures.13d,eSoaking of the Fe(TCNQ)(4,4′-bpy) MOFs in benzene, toluene, aniline and nitrobenzene was carried out with degassed solvents under a nitrogen atmosphere. Powder X-ray diffraction measurements were performed with a BRUKER D8-Focus Bragg-Brentano X-ray Powder Diffractometer equipped with an CuKα radiation source (λ = 1.5406 Å, 40 kV and 40 mA). The samples for magnetic measurements were prepared by sealing crushed crystals with a minimum volume of the corresponding solvents in quartz tubes.
Synthesis of Fe(TCNQ)(DMF)2·2DMF
A solution of 0.2 mmol of Fe(SO4)2·7H2O, 0.5 mmol of Li(OAc)·2H2O and 20 mg of ascorbic acid in methanol was layered on a solution of 0.2 mmol of H2TCNQ in (N,N-dimethylformamide (DMF). Yellowish green crystals formed in two days. IR: ν(CN) 2196.7, 2134.5 cm−1; ν(CO) 1639.7 cm−1.Conclusions
The properties of TCNQ-based magnetic MOF materials were investigated by magnetic, structural and infrared studies. The magnetic properties of Fe(TCNQ)(4,4′-bpy) MOFs can be modulated but altering the polarity and the size of the aromatic guest molecules. In addition, investigation of the magnetic properties of this series of magnetic MOFs has revelaed that the TCNQ-dianion as a bridging ligand for the mediation of magnetic coupling is a promising candidate for the synthesis of magnetic MOF materials. Structural evidence was obtained to support the conclusion that significant steric and electrostatic interactions between the polar nitrobenzene and the negatively charged TCNQ2− species occurs in the frameworks. The 2-D model compound Fe(TCNQ)(DMF)2·2DMF was synthesized and the magnetic studies suggest structural distortion is responsible for the transition to canted spin states. A comparison with isostructural Mn(II) analogs of the MOFs indicate that single-ion magnetic anisotropy is crucial for the presence of canted spin states in the Fe-TCNQ-based MOFs. The magnetic phase transitions are due to electrostatic and steric interactions between the guest molecules and the host frameworks. The opportunity to modulate magnetic properties by the interaction of TCNQ dianions in framework solids with aromatic small molecules of different polarity and steric hindrance make them promising for applications in molecule recognition and sensors.Acknowledgements
This material is based on work supported by the U.S. Department of Energy, Office of Basic Energy Sciences, Division of Materials Sciences and Engineering under Award DE-SC0012582. MFBR thanks Secretaria de Ciencia y Tecnologia del Distrito Federal (SECITI). The authors thank Dr John Bacsa for his helpful discussions and Mr Toby Woods for collecting some of the X-ray diffraction data. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. ChemMatCARS Sector 15 is supported by the National Science Foundation under grant number NSF/CHE-1346572.Notes and references
- (a) J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869 CrossRef CAS PubMed; (b) R. B. Getman, Y.-S. Bae, C. E. Wilmer and R. Q. Snurr, Chem. Rev., 2012, 112, 703 CrossRef CAS PubMed; (c) M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196 CrossRef CAS PubMed; (d) L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105 CrossRef CAS.
- (a) D. Feng, W.-C. Chung, Z. Wei, Z.-Y. Gu, H.-L. Jiang, Y.-P. Chen, D. J. Darensbourg and H.-C. Zhou, J. Am. Chem. Soc., 2013, 135, 17105 CrossRef CAS PubMed; (b) M. J. Katz, J. E. Mondloch, R. K. Totten, J. K. Park, S. T. Nguyen, O. K. Farha and J. T. Hupp, Angew. Chem., Int. Ed., 2014, 53, 497 CrossRef CAS PubMed.
- (a) T. C. Narayan, T. Miyakai, S. Seki and M. Dincă, J. Am. Chem. Soc., 2012, 134, 12932 CrossRef CAS PubMed; (b) C. H. Hendon, D. Tiana and A. Walsh, Phys. Chem. Chem. Phys., 2012, 14, 13120 RSC; (c) A. A. Talin, A. Centrone, A. C. Ford, M. E. Foster, V. Stavila, P. Haney, R. A. Kinney, V. Szalai, F. El Gabaly, H. P. Yoon, F. Léonard and M. D. Allendorf, Science, 2014, 343, 66 CrossRef CAS PubMed; (d) L. Sun, C. H. Hendon, M. A. Minier, A. Walsh and M. Dinca, J. Am. Chem. Soc., 2015, 137, 6164 CrossRef CAS; (e) T. Kambe, R. Sakamoto, K. Hoshiko, K. Takada, M. Miyachi, J.-H. Ryu, S. Sasaki, J. Kim, K. Nakazato, M. Takata and H. Nishihara, J. Am. Chem. Soc., 2013, 135, 2462 CrossRef CAS PubMed; (f) D. Sheberla, L. Sun, M. A. Blood-Forsythe, S. Er, C. R. Wade, C. K. Brozek, A. Aspuru-Guzik and M. Dinca, J. Am. Chem. Soc., 2014, 136, 8859 CrossRef CAS PubMed.
- (a) G. Lorusso, J. W. Sharples, E. Palacios, O. Roubeau, E. K. Brechin, R. Sessoli, A. Rossin, F. Tuna, E. J. L. McInnes, D. Collison and M. Evangelisti, Adv. Mater., 2013, 25, 4653 CrossRef CAS PubMed; (b) M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353 RSC.
- (a) T.-H. Chen, I. Popov, W. Kaveevivitchai and O. Š. Miljanić, Chem. Mater., 2014, 26, 4322 CrossRef CAS; (b) F. A. Almeida Paz, J. Klinowski, S. M. F. Vilela, J. P. C. Tome, J. A. S. Cavaleiro and J. Rocha, Chem. Soc. Rev., 2012, 41, 1088 RSC.
- (a) T. B. Faust and D. M. D'Alessandro, RSC Advances, 2014, 4, 17498–17512 RSC; (b) H. Miyasaka, Acc. Chem. Res., 2013, 46, 248 CrossRef CAS PubMed; (c) C. Avendano, Z. Zhang, A. Ota, H. Zhao and K. R. Dunbar, Angew. Chem., Int. Ed., 2011, 50, 6543 CrossRef CAS PubMed; (d) A. Nafady, A. P. O'Mullane and A. M. Bond, Coord. Chem. Rev., 2014, 268, 101 CrossRef CAS; (e) A. Nafady, T. H. Le, N. Vo, N. L. Haworth, A. M. Bond and L. L. Martin, Inorg. Chem., 2014, 53, 2268–2275 CrossRef CAS PubMed; (f) B. F. Abrahams, R. W. Elliott, T. A. Hudson and R. Robson, Cryst. Growth Des., 2013, 13, 3018 CrossRef CAS; (g) S. Shimomura, S. Horike, R. Matsuda and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 10990 CrossRef CAS PubMed; (h) S. Horike, M. Sugimoto, K. Kongpatpanich, Y. Hijikata, M. Inukai, D. Umeyama, S. Kitao, M. Seto and S. Kitagawa, J. Mater. Chem. A, 2013, 1, 3675 RSC; (i) B. F. Abrahams, R. W. Elliott and R. Robson, Aust. J. Chem., 2014, 67, 1871 CrossRef CAS; (j) S. S. Park, E. R. Hontz, L. Sun, C. H. Hendon, A. Walsh, T. Van Voorhis and M. Dincă, J. Am. Chem. Soc., 2015, 137, 1774–1777 CrossRef CAS PubMed.
- (a) A. Aspuru-Guzik, A. D. Dutoi, P. J. Love and M. Head-Gordon, Science, 2005, 309, 1704 CrossRef CAS PubMed; (b) M. J. Graham, J. M. Zadrozny, M. Shiddiq, J. S. Anderson, M. S. Fataftah, S. Hill and D. E. Freedman, J. Am. Chem. Soc., 2014, 136, 7623 CrossRef CAS; (c) M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789 CrossRef CAS PubMed.
- (a) R. A. Heintz, H. Zhao, X. Ouyang, G. Grandinetti, J. Cowen and K. R. Dunbar, Inorg. Chem., 1999, 38, 144 CrossRef CAS; (b) H. Zhao, J. M. J. Bazile, J. R. Galán-Mascarós and K. R. Dunbar, Angew. Chem., Int. Ed., 2003, 42, 1015 CrossRef CAS PubMed; (c) N. Lopez, H. Zhao, A. Ota, A. V. Prosvirin, E. W. Reinheimer and K. R. Dunbar, Adv. Mater., 2010, 22, 986 CrossRef CAS PubMed; (d) M. Ballesteros-Rivas, A. Ota, E. Reinheimer, A. Prosvirin, J. Valdes-Martinez and K. R. Dunbar, Angew. Chem., Int. Ed., 2011, 50, 9703 CrossRef CAS PubMed; (e) X. Zhang, Z. Zhang, H. Zhao, J. G. Mao and K. R. Dunbar, Chem. Commun., 2014, 50, 1429 RSC; (f) Z. Zhang, H. Zhao, M. M. Matsushita, K. Awaga and K. R. Dunbar, J. Mater. Chem. C, 2014, 2, 399 RSC; (g) H. Kojima, Z. Zhang, K. R. Dunbar and T. Mori, J. Mater. Chem. C, 2013, 1, 1781 RSC; (h) Z. Zhang, H. Zhao, H. Kojima, T. Mori and K. R. Dunbar, Chem. – Eur. J., 2013, 19, 3348 CrossRef CAS PubMed; (i) Z. X. Wang, X. Zhang, Y. Z. Zhang, M. X. Li, H. Zhao, M. Andruh and K. R. Dunbar, Angew. Chem., Int. Ed., 2014, 11567 CrossRef CAS PubMed.
- (a) L. L. Cheruiyot, R. J. Crutchley, L. K. Thompson, J. E. Greedan and G. Liu, Can. J. Chem., 1995, 73, 573 CrossRef CAS; (b) W. Kosaka, T. Morita, T. Yokoyama, J. Zhang and H. Miyasaka, Inorg. Chem., 2015, 54, 1518 CrossRef CAS PubMed; (c) E. Ruiz, A. Rodríguez-Fortea and S. Alvarez, Inorg. Chem., 2003, 42, 4881 CrossRef CAS PubMed.
- M. R. Saber, A. V. Prosvirin, B. F. Abrahams, R. W. Elliott, R. Robson and K. R. Dunbar, Chem. – Eur. J., 2014, 20, 7593 CrossRef CAS PubMed.
- (a) P. Dechambenoit and J. R. Long, Chem. Soc. Rev., 2011, 40, 3249 RSC; (b) Z.-M. Hao and X.-M. Zhang, Dalton Trans., 2011, 40, 2092 RSC; (c) M. Ohba, K. Yoneda and S. Kitagawa, CrystEngComm, 2010, 12, 159 RSC; (d) O. Kahn, J. Larionova and J. V. Yakhmi, Chem. – Eur. J., 1999, 5, 3443 CrossRef CAS.
- N. Lopez, H. Zhao, A. V. Prosvirin, A. Chouai, M. Shatruk and K. R. Dunbar, Chem. Commun., 2007, 4611 RSC.
- (a) S. Shimomura, R. Matsuda, T. Tsujino, T. Kawamura and S. Kitagawa, J. Am. Chem. Soc., 2006, 128, 16416 CrossRef CAS PubMed; (b) S. Shimomura, M. Higuchi, R. Matsuda, K. Yoneda, Y. Hijikata, Y. Kubota, Y. Mita, J. Kim, M. Takata and S. Kitagawa, Nat. Chem., 2010, 2, 633 CrossRef CAS PubMed; (c) S. Shimomura, R. Matsuda and S. Kitagawa, Chem. Mater., 2010, 22, 4129 CrossRef CAS; (d) S. Shimomura, N. Yanai, R. Matsuda and S. Kitagawa, Inorg. Chem., 2010, 50, 172 CrossRef PubMed; (e) B. F. Abrahams, R. W. Elliott, T. A. Hudson and R. Robson, Cryst. Growth Des., 2010, 10, 2860 CrossRef CAS.
- C. Kittel, Introduction to solid state physics, Wiley, 1986 Search PubMed.
- M. E. Lines, J. Phys. Chem. Solids, 1970, 31, 101 CrossRef CAS.
- (a) S. Triki, F. Thetiot, J.-R. Galan-Mascaros, J. S. Pala and K. R. Dunbar, New J. Chem., 2001, 25, 954 RSC; (b) S. R. Marshall, C. D. Incarvito, J. L. Manson, A. L. Rheingold and J. S. Miller, Inorg. Chem., 2000, 39, 1969 CrossRef CAS.
- (a) J. L. Manson, C. D. Incarvito, A. L. Rheingold and J. S. Miller, J. Chem. Soc., Dalton Trans., 1998, 3705 RSC; (b) J. L. Manson, A. M. Arif, C. D. Incarvito, L. M. Liable-Sands, A. L. Rheingold and J. S. Miller, J. Solid State Chem., 1999, 145, 369 CrossRef CAS; (c) Y. Rodriguez-Martín, C. Ruiz-Pérez*, J. n. Sanchiz, F. Lloret* and M. Julve, Inorg. Chim. Acta, 2001, 318, 159 CrossRef.
- A. V. Prosvirin, H. Zhao and K. R. Dunbar, Inorg. Chim. Acta, 2012, 389, 118 CrossRef CAS PubMed.
- D. S. Acker and W. R. Hertler, J. Am. Chem. Soc., 1962, 84, 3370 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallography details and additional magnetic data. CCDC 1414461–1414463 and 1414477. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00128e |
| This journal is © the Partner Organisations 2015 |