
Graphene oxide and reduced graphene oxide hybrids with spin crossover iron(III) complexes†
Yusuke
Murashima
a,
Mohammad Razaul
Karim
b,
Naoto
Saigo
a,
Hiroshi
Takehira
a,
Ryo
Ohtani
a,
Masaaki
Nakamura
a,
Michio
Koinuma
a,
Leonard F.
Lindoy
c,
Keita
Kuroiwa
d and
Shinya
Hayami
*ae
aGraduate School of Science and Technology, Kumamoto University, 2-39-1 Kurokami, Chuo-ku, Kumamoto 860-8555, Japan. E-mail: hayami@sci.kumamoto-u.ac.jp
bDepartment of Chemistry, School of Physical Sciences, Shahjalal University of Science & Technology, Sylhet-3114, Bangladesh
cSchool of Chemistry, The University of Sydney, NSW 2006, Australia
dDepartment of Nanoscience, Faculty of Engineering, Sojo University, 4-22-1 Ikeda, Nishi-ku, Kumamoto 860-0082, Japan
eInstitute of Pulsed Power Science (IPPS), Kumamoto University, 2-39-1 Kurokami, Chuo-ku, Kumamoto 860-8555, Japan
First published on 23rd July 2015
Abstract
Graphene (rGO) based hybrid materials exhibiting electrical conductivity and spin crossover (SCO) behavior are reported. The non-conductive [Fe(qnal)2]nGO (1·GO) and [Fe(qsal)2]nGO (2·GO) hybrids have been prepared by employing the electrostatic interaction between the negatively charged graphene oxide (GO) nanosheet and the respective iron(III) complex cations in [Fe(qnal)2]+Cl− and [Fe(qsal)2]+Cl−. The conductive [Fe(qnal)2]nrGO (1·rGO) and [Fe(qsal)2]nrGO (2·rGO) hybrids were obtained by thermal reduction of 1·GO and 2·GO. 1·GO and 1·rGO exhibit SCO behavior, and 1·rGO also shows a light-induced excited spin state trapping (LIESST) effect. Thus, in 1·rGO the electrical conductivity of rGO and the SCO behavior of [Fe(qnal)2]+ coexist in a single structure. We propose that the observed cooperativity for the rGO nanosheet-bound iron(III) [Fe(qnal)2]+ SCO material occurs through formation of large domains via π–π stacking between the graphene skeleton and the [Fe(qnal)2]+ cations.
Introduction
Reported strategies for combining electrical conductivity with magnetism in hybrid materials involve the combination of magnetic ingredients with a conductive organic/inorganic platform by chemical bond formation, columbic attachment or layer by layer assembly of the functional species within a porous polymeric framework.1,2 In particular, aggregation through non-covalent self-assembly of multi-components to form stable functional molecular materials has received wide interest, as the properties of the components are being preserved largely.3 The layering of individual functional materials to form multifunctional hybrids has been successfully adopted for obtaining coalescence between ferromagnetism and metallic conductivity, paramagnetism and superconductivity and ferromagnetism and superconductivity.4–6 In most of these systems magnetic nanoparticles are physically trapped in an appropriate conductive substrate either by stable sandwich formation or through the trapping of the functional components in a folded dispersion phase.7 Due to the near homogeneous height profile of the conductive mesoporous dispersion phase and its limited interface with the magnetic species, magnetic ordering is largely retained, resulting in the preservation of magnetic properties. In this context, there has been considerable interest in using reduced graphene oxide (rGO) as a thin, conductive two dimensional dispersed phase.8 An additional advantage of using rGO is the possibility for RKKY exchange between graphene's p-orbital based electrons and the magnetic entities, which could further enhance magnetic ordering.9,10 However, there are inherent difficulties in embedding spin crossover (SCO) materials in such a conductive matrix.SCO represents magnetic behavior involving the transition of an electron (or electrons) between high spin (HS) and low spin (LS) metal states and is commonly observed for first row transition metal complexes with 3dn (n = 4–7) electronic configurations when they are exposed to external stimuli such as temperature, pressure, magnetic field or light.11 Depending on the extent of cooperativity between the molecules, solid state SCO behavior can be gradual or abrupt. Cooperativity, reflecting the presence of significant intermolecular interaction between the SCO molecules, is highly desirable as it induces abrupt SCO behavior (all the molecules in a sample undergo their spin transitions simultaneously) giving rise to hysteresis loops. Such behavior is important in terms of possible applications such as data storage, magnetic switching and optical devices. However, achieving such cooperativity among SCO molecules is not easy, as achieving simultaneous electron transitions across all molecules in a non-homogeneous system (with respect to ligand field, spatial arrangement or dimensionally controlled alignment) is difficult. Considering the role of intermolecular interactions in inducing spin transitions, it was proposed theoretically (and experimentally confirmed) that cooperativity is enhanced by designing polymeric structures, in which the active sites are linked to each other through chemical bridges.12–17 The SCO behavior of a complex is well documented to be affected significantly by ligand modification or by interaction of the complex with other materials. However, the generation of an additional functionality (e.g. electrical conductivity) in a SCO system by direct elaboration of the system lies beyond theoretical prediction at the present time.
Even though the design of SCO materials with other desired chemical or physical properties has long been a major focus in hybrid materials research,18,19 it has only been possible to combine magnetism and SCO in recent times,20,21 with very few materials being developed that combine SCO and electrical conductivity in a single platform.22–28 In most cases discrete entities are combined by Coulombic attraction (rather than the covalent linkage). For example, the use of π-stacking and electrostatic attraction resulted in conductive SCO hybrids based on the redox active [Ni(dmit)2]− anion and the spin crossover [Fe(sal2-trien)]+ complex cation.29 Another example exploited the synergism between the electrical conductivity of stacks of [Ni(dmit)2]− in electrocrystallized [Fe(qsal)2][Ni(dmit)2]3·CH3CN·H2O and the SCO of [Fe(qsal)2]+.30 Although there exist reports of cationic complex SCO behaviour coupled with anionic metal complex conduction, no hybrids based on rGO or any other purely organic framework are known.
In view of the above, we initially considered combining the SCO nature of [Fe(qsal)2]+ or [Fe(qnal)2]+ (qsal and qnal are the abbreviations of the deprotonated Hqsal (N-(8-quinolyl)salicylaldimine)) and Hqnal (1-((8-quinolinylimino)methyl)-2-naphthalenol), respectively as shown in Fig. 1) with the conductivity of rGO. rGO is similar to graphene, which is a well documented electronic conductor31 and [Fe(qsal)2]+ and [Fe(qnal)2]+ are both reported SCO entities.32,33 The SCO behavior of [Fe(qsal)2]+ and [Fe(qnal)2]+ in the presence of various counter anions has been studied extensively while both rGO and GO have perfect two dimensional structures at room temperature.34
 | ||
| Fig. 1 Structures of the ligands, Hqsal and Hqnal. | ||
In the present study, the combination of [Fe(qsal)2]+ and [Fe(qnal)2]+ with the negatively charged GO resulted in the formation of [Fe(qnal)2]nGO (1·GO) and [Fe(qsal)2]nGO (2·GO) as non-conductive GO hybrids (Scheme 1). However, on thermal reduction, these GO hybrids yielded conductive graphene hybrids as [Fe(qnal)2]nrGO (1·rGO) and [Fe(qsal)2]nrGO (2·rGO). In addition, 1·GO and 1·rGO each maintain SCO behavior. Furthermore, 1·rGO also shows a light-induced excited spin state trapping (LIESST) effect. The direct synergy between the electrical conductivity of rGO and the respective spin conversions is shown to involve the cooperativity between the SCO molecules embedded on the rGO nanosheets. This result opens a new window for the generation of rGO based hybrid materials for switching and other light driven optical device applications.
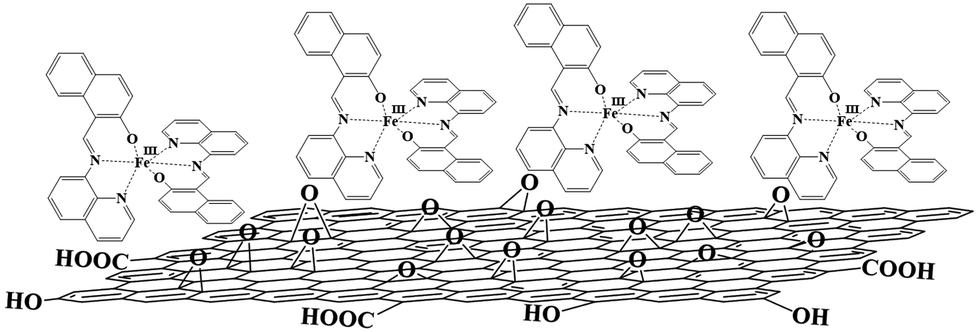 | ||
| Scheme 1 Scheme for the electrostatic interaction between the positively charged [Fe(qnal)2]+ and the negatively charged GO, and [Fe(qsal)2]+ also is proposed to bind to GO by the same interaction. | ||
Experimental
Graphene oxide synthesis
Graphene oxide was prepared by a modified Hummers’ method we reported elsewhere.35 0.50 g of graphite powder (95%) and 0.5 g of NaNO3 were mixed in a 200 mL flask placed into an ice bath. 11.5 mL of conc. H2SO4 was added and the mixture was stirred for 30 min at 0 °C. 1.5 g of KMnO4 was added slowly with stirring. The mixture was then stirred for 1 h at 35 °C. 23 mL of distilled water was slowly added and the mixture was stirred for 30 min at 95 °C. The reaction was terminated by adding 100 mL of distilled water followed by 3 mL of 30% H2O2 solution. The mixture was cooled and centrifuged at 3000 rpm to remove the supernatant liquid. The remaining solid mixture was washed with 5% HCl acid (1 time) and distilled water (3 times). The precipitate was dried at 80 °C overnight to yield graphite oxide (GtO). 0.2 g of GtO was added into 200 mL deionized water and sonicated for 2 h. This dispersion was centrifuged at 4000 rpm for 1 h and the supernatant liquid was called graphene oxide (GO) dispersion (Fig. S1†).Synthesis of [Fe(qsal)2]Cl
A solution of 8-aminoquinoline (1.44 g, 10 mmol) and salicylaldehyde (1.22 g, 10 mmol) in methanol (10 mL) was heated to reflux for 1 h. The obtained orange solution was cooled and FeCl3·6H2O (1.35 g, 5 mmol) was added with vigorous stirring. Finally, triethylamine (1.01 g, 10 mmol) was added slowly to the solution. The reaction mixture was stirred at room temperature for 1 h to yield a black precipitate.36Synthesis of [Fe(qnal)2]Cl
A solution of 8-aminoquinoline (0.28 g, 2 mmol) in methanol (20 mL) and 2-hydroxy-1-naphthaldehyde (0.34 g, 2 mmol) in methanol (10 mL) was stirred for 1 h in order to produce Hqnal. Meanwhile, FeCl3 (0.16 g, 1 mmol) was dissolved in absolute methanol (5 mL) containing 2,2′-dimethoxy-propane (2 mL) and warmed at 40 °C for 30 min, and was added to the Hqnal in methanol. The micro-crystalline black product was collected by suction and dried in a vacuum.32Synthesis of GO–SCO hybrids
GO–SCO hybrids, 1·GO and 2·GO, were directly precipitated from a mixture of GO dispersion (200 mL) and iron(III) complexes ([Fe(qsal)2]Cl and [Fe(qnal)2]Cl) solution (200 mg in 200 mL methanol) at 25 °C for 24 h. The hybrids were washed with water (3 times) with centrifugation (4800 rpm), collected by membrane filter and dried in a vacuum (Fig. S1†).Synthesis of rGO–SCO hybrids
rGO–SCO hybrids, 1·rGO and 2·rGO, were prepared by thermal reduction of GO–SCO hybrids. The powder of GO–SCO hybrids was heated under argon at 120 °C for 24 h (Fig. S1†).Physical measurements
To perform transmission electron microscopy (TEM; JEOL, 2000FX, 200 kV), one drop of the aqueous nanosheet suspension was deposited on a holey carbon film. A micro Raman spectrometer (NRS-3100, Jasco, Japan) with a 532 nm excitation source at room temperature was used to collect the Raman spectra. XPS (Thermo Scientific, Sigma Probe) was used to analyze the surface of GO nanosheets. A monochromatized X-ray source (Al Kα, hν = 1486.6 eV) was used for XPS. In this measurement, a Pt substrate (GO/Pt film) was used to determine the fermi level. All measurements were done in a vacuum better than 10−7 Pa. Emitted electrons were detected by using a hemispherical energy analyzer equipped with six channeltrons. The powder X-ray diffraction (XRD) patterns were recorded on a Rigaku X-ray diffractometer (RAD-2A with a 2.0 kW CuKα X-ray).Temperature dependence of the electrical resistivity of a pelt of GO–SCO and rGO–SCO hybrids was measured by using a nanovoltmeter (Keithley 2182A Digital Nanovoltmeter) using a four-terminal method in the temperature range of 150–300 K. The conductivities along the surface of GO–SCO hybrids and rGO–SCO hybrids were measured on a glass substrate by a two electrode system.
The magnetic susceptibilities for GO–SCO and rGO–SCO hybrids between 2 K and 400 K were measured with a superconducting quantum interference device (SQUID) magnetometer (Quantum Design MPMSXL-5) in an external field of 1 T.
The Mössbauer spectra were recorded with a Wissel MVT-1000 Mössbauer spectrometer with a 57Co/Rh source in the transition mode. All isomer shifts are given relative to α-Fe at room temperature. Measurements at low temperature were performed with a closed-cycle helium refrigerator cryostat (Iwatani Co., Ltd).
Results and discussion
The morphologies of the GO and its hybrid derivatives 1·GO and 2·GO are illustrated in the TEM images shown in Fig. 2(a), (b) and (d). The folded GO nanosheet is darkened in its 1·GO and 2·GO hybrids. Following reduction, the randomly distributed dark spots in the former hybrids become denser and more widespread in 1·rGO and 2·rGO due to nanoparticles formed on the rGO surface following coagulation of the respective iron complexes (Fig. 2(c) and (e)). In 1·GO and 2·GO the binding of cations on the negatively charged GO nanosheet is stabilized by charge balance and further coagulation of the metal complexes is inhibited. However, on reduction to rGO, the charge disappears and the hybrids are stabilized by encapsulation of coagulated metal complexes giving rise to the formation of dense and more defined spots. It is also proposed that the secondary positive charge on the metal complex cations is partly balanced by Cl− anions. During reduction electron acceptance also likely results in some secondary charge balance, which aids coagulation. That is, the GO nanosheets trap the SCO molecules by electrostatic interaction (Scheme 1) and after thermal reduction the coagulated mass is sandwiched within the 2D carbon matrix of rGO in each case. | ||
| Fig. 2 TEM images for (a) GO, (b) 1·GO, (c) 1·rGO, (d) 2·GO and (e) 2·rGO. | ||
The PXRD pattern of GO shows a characteristic peak at 11.27° (2θ), with a d spacing of 7.83 Å signifying the presence of intercalated oxygen atoms positioned at various oxygenated functional sites (Fig. S2†).37 Due to the accommodation of [Fe(qnal)2]+ and [Fe(qnsl)2]+ by the GO nanosheet, the respective d spacing increases to 9.16 and 8.89 Å with the peak positions (2θ) occurring at 9.65° and 9.64°, respectively. When GO is reduced to rGO, the coagulated mass of the respective iron(III) complexes is encapsulated by folded layers of rGO, with the characteristic peak for the oxygenated sites no longer present. In the case of 2·rGO, a new shallow peak in the high angle region is seen, which is in accord with the lowering of the interlayer distance due to the removal of epoxy sites from some regions of the GO nanosheet during reduction. However, for 1·rGO, the corresponding shallow peak is not so clear.
The Raman spectra of GO, 1·GO and 2·GO are compared in Fig. S3.† The chemical change corresponding to the conversion of GO to rGO and the columbic interaction between the GO nanosheet and [Fe(qnal)2]+ is reflected by the change in the position of the G band and relative heights of the D (1357 cm−1) and G (1591 cm−1) bands; the D and G bands correspond to the breathing mode of A1g and in-plane bond stretching motion of pairs of sp2 C atoms (E2g) mode, respectively.38,39 The G band position for GO shifts to 1593 cm−1 in 1·GO and then to 1589 cm−1 on reduction of 1·GO to 1·rGO. The slight hardening (shifting of peaks to a high frequency region) during the formation of 1·GO from GO implies a change in the electronic environment due to the resonance through the π–π interaction between the organic framework of the iron complex and the GO nanosheet. In this context, it is noted that the presence of an electron rich heteroatom or chemical modification has also been reported to result in such hardening.40,41 While 1·GO is changed to 1·rGO, the slight softening (shifting of peaks to a low frequency region) is an expected consequence of the π–π interaction being lowered. The peak ratio (ID/IG) increases from 0.87 in GO to 0.98 in 1·GO and 1.00 in 1·rGO. The ID/IG is inversely proportional to the extent of the sp2 C domain. Therefore, an increase in this value signifies the fragmentation of large sp2 islands of GO during the metal complex attachment and subsequent reduction.42,43 In addition, the D band intensity is related to the extent of defects in the GO plane.44 These defects arise from the metal complex deposition on the GO nanosheet. In the case of 2·GO, the change in Raman parameters is very similar to that discussed above. Here, the G band positions are 1591, 1595 and 1599 cm−1 for GO, 2·GO and 2·rGO, respectively. The respective ID/IG ratios are, 0.87, 0.91 and 1.04.
GO exhibits two characteristic peaks near 285 and 287 eV in its XPS spectra (Fig. 3). Oxygen functional groups in GO were present in the form of epoxide (–O–) and hydroxyl (–OH) groups with XPS peak positions in the range 286.8–287.0 eV as well as carbonyl (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), and carboxyl (–COOH) groups between 287.8 and 288.0 eV and 289.0 and 289.3 eV, respectively.45,46 The peak for oxygenated C 1s is narrower and higher compared to the C 1s peak for unoxidized carbon sites (–C–C– and –CC–). The relative amounts of –C–O, –C–O–C–, –CO and OC–O– functional sites are 3.9, 32.3, 6.3 and 7.5%, respectively. The ratio of area under the peak for oxygenated C 1s to the area of oxygenated and unoxygenated C 1s carbon sites reveals that the carbon to oxygen ratio is around 7
O), and carboxyl (–COOH) groups between 287.8 and 288.0 eV and 289.0 and 289.3 eV, respectively.45,46 The peak for oxygenated C 1s is narrower and higher compared to the C 1s peak for unoxidized carbon sites (–C–C– and –CC–). The relative amounts of –C–O, –C–O–C–, –CO and OC–O– functional sites are 3.9, 32.3, 6.3 and 7.5%, respectively. The ratio of area under the peak for oxygenated C 1s to the area of oxygenated and unoxygenated C 1s carbon sites reveals that the carbon to oxygen ratio is around 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 and comply with a previous report.47 In Fig. 3(a) the carbon and oxygen contents are changed in 1·GO as 72.1 and 24.79% and in 1·rGO as 75.55 and 21.48%. The carbon content increases due to the attachment of [Fe(qnal)2]+ on GO and rGO. For reduction the carbon content increases further due to the breaking of epoxy sites and removal of other oxygen functional groups. The N, Fe, and Cl contents in 1·GO and 1·rGO are 1.97, 0.48, 0.5 and 2.05, 0.45, 0.35%, respectively. The N:Fe:Cl ratio is almost 4:1:1 in each sample confirming the presence of the Cl− anion and in keeping with the undissociated [Fe(qnal)2]+ ion being present on the GO nanosheet. We propose that the Cl− anion becomes attached to GO through some non-specific adsorption related to achieving charge balance. On reduction, the decrease in the peak height near 287 eV is in accord with the destruction of some epoxy and hydroxyl groups having occurred. However, following reduction these peaks have not completely disappeared, thus indicating that some epoxy and hydroxyl groups remain intact. 2·GO and 2·rGO hybrids also exhibited close to similar behavior (Fig. 3(b)).
:3 and comply with a previous report.47 In Fig. 3(a) the carbon and oxygen contents are changed in 1·GO as 72.1 and 24.79% and in 1·rGO as 75.55 and 21.48%. The carbon content increases due to the attachment of [Fe(qnal)2]+ on GO and rGO. For reduction the carbon content increases further due to the breaking of epoxy sites and removal of other oxygen functional groups. The N, Fe, and Cl contents in 1·GO and 1·rGO are 1.97, 0.48, 0.5 and 2.05, 0.45, 0.35%, respectively. The N:Fe:Cl ratio is almost 4:1:1 in each sample confirming the presence of the Cl− anion and in keeping with the undissociated [Fe(qnal)2]+ ion being present on the GO nanosheet. We propose that the Cl− anion becomes attached to GO through some non-specific adsorption related to achieving charge balance. On reduction, the decrease in the peak height near 287 eV is in accord with the destruction of some epoxy and hydroxyl groups having occurred. However, following reduction these peaks have not completely disappeared, thus indicating that some epoxy and hydroxyl groups remain intact. 2·GO and 2·rGO hybrids also exhibited close to similar behavior (Fig. 3(b)).
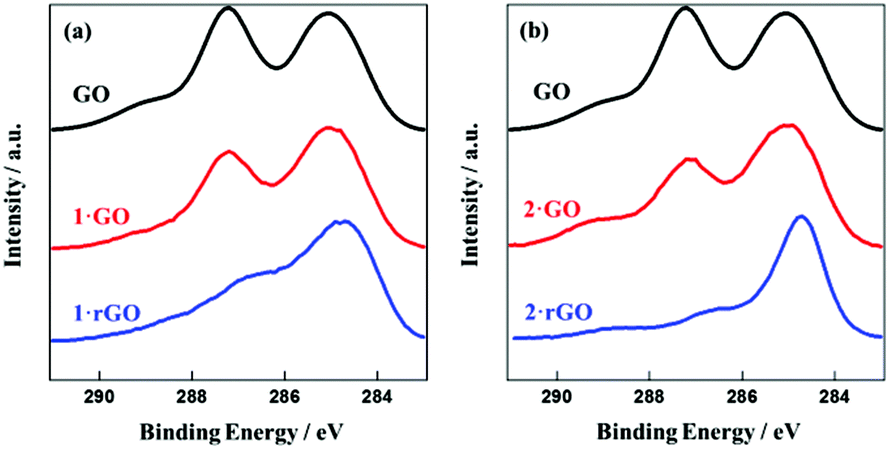 | ||
| Fig. 3 XPS spectra for (a) GO (black), 1·GO (red) and 1·rGO (blue), and (b) GO (black), 2·GO (red) and 2·rGO (blue). | ||
While GO is a well known insulator with respect to electron conduction, conductivity is recovered after reduction.48 Temperature dependent electrical resistivities for 1·rGO and 2·rGO were measured in the temperature range of 150–300 K employing a HUSO HECS 9065 conductometer using the conventional four-probe method (Fig. S4†). Both hybrids show semiconducting behavior. For 1·rGO the resistivity at 150 K is 2.70 × 105 Ω cm and this reduces exponentially with temperature. At 300 K the resistivity is almost zero. For 2·rGO a similar trend is observed with the resistivity being 95 and 0 Ω cm at 150 and 300 K, respectively. The conductive nature of all the hybrids was further probed by observing the current (I)–voltage (V) curves for the hybrids before and after thermal reduction. The I–V curves are presented in Fig. 4. The unreduced hybrids show zero conductivity regardless of the applied voltage. However, for 1·rGO and 2·rGO the current increases linearly with voltage. At 1 V, the electron conductivities for 1·rGO and 2·rGO are 18 and 680 μA, respectively.
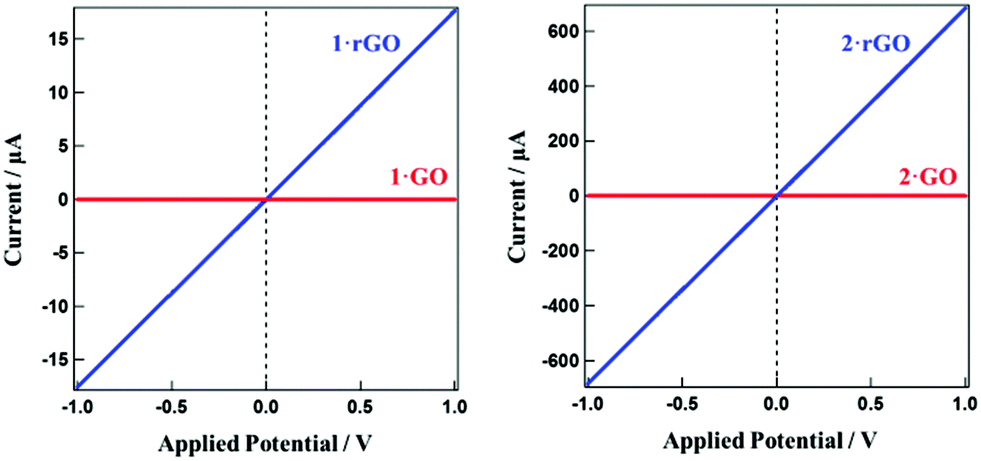 | ||
| Fig. 4 The related I–V curve showing the conductivity of the hybrids for (a) 1·GO and 1·rGO, and (b) 2·GO and 2·rGO. | ||
The χgT value versus temperature behavior for 1·GO is shown in Fig. S5.† The value of χgT for 1·GO remains constant at 1.65 × 10−3 cm3 K g−1 from 2 to 180 K. From 180 to 260 K, χgT rises gradually, then follows a steeper increase beyond 260 K. At 400 K the χgT value reaches 2.28 × 10−3 cm3 K g−1. For 1·rGO, χgT is 1.9 × 10−3 cm3 K g−1 at 10 K. A gradual increase with constant steep is then observed, with the χgT value reaching 2.5 × 10−3 cm3 K g−1 at 400 K. Both 1·GO and 1·rGO displays clear evidence for SCO behavior. However, as both GO and rGO are nanostructured polymers, their size and shape cannot be controlled, and the exact molecular formula (or weight) of the GO hybrids is unable to be calculated. Therefore, in place of emu K mol−1, magnetic susceptibility is expressed in terms of cm3 K g−1. Though the temperature dependent spin transition is very clear, the fraction of iron(III) in the low and high spin states before and after the spin transition in 1·GO and 1·rGO is unable to be calculated from the susceptibility measurement. We also estimated the high-spin (HS) and low-spin (LS) ratio in SCO by means of 57Fe Mössbauer spectra. The temperature dependence of the Mössbauer spectra of 1·GO is presented in Fig. 5. The black dots represent the observed spectra while the blue and red lines represent the simulated LS and HS curves, respectively. The black line represents the sum of the simulated LS and HS curves. The Mössbauer parameters at 300 K for 1·GO are as follows. HS area: AHS = 61%, isomer shift (I.S.) = 0.38 mm s−1, quadrupole splitting (Q.S.) = 0.76 mm s−1, and LS area: ALS = 39% I.S. = 0.17 and Q.S. = 2.32. At 5 K, AHS = 36.0%, I.S. = 0.51, Q.S. = 0.90, and ALS = 64%, I.S. = 0.22 and Q.S. = 2.55. The HS fraction (γHS) increases from 0.36 to 0.61 on heating (5–300 K). In the case of 1·rGO, the Mössbauer parameters at 300 K are as follows. AHS = 65%, I.S. = 0.38 mm s−1 and Q.S. = 0.77 mm s−1, and ALS = 35%, I.S. = 0.17 mm s−1 and Q.S. = 2.24 mm s−1. At 5 K, AHS = 41%, I.S. = 0.51 mm s−1, Q.S. = 0.89 mm s−1, and ALS = 59%, I.S. = 0.21 mm s−1, Q.S. = 2.52 mm s−1. γHS increases from 0.41 to 0.65 on heating (5–300 K). The Mössbauer spectra therefore confirm the temperature dependent SCO of 1·GO and 1·rGO. The other hybrids 2·GO and 2·rGO were also studied by SQUID and Mössbauer spectra, but exhibited no SCO behavior (Fig. S5 and S6†). These differences result from the different domain sizes of [Fe(qnal)2]+ and [Fe(qsal)2]+ metal complex cations on GO and rGO. The cation [Fe(qnal)2]+ has a larger π-conjugated ligand that strongly interacts with GO and rGO. As a result, the domain size of [Fe(qnal)2]+ is large on GO and rGO, 1·GO and 1·rGO show SCO behavior. On the other hand, the domain size of [Fe(qsal)2]+ is smaller because of a smaller π-conjugated ligand on GO and rGO, and 2·GO and 2·rGO don't show the SCO behavior.
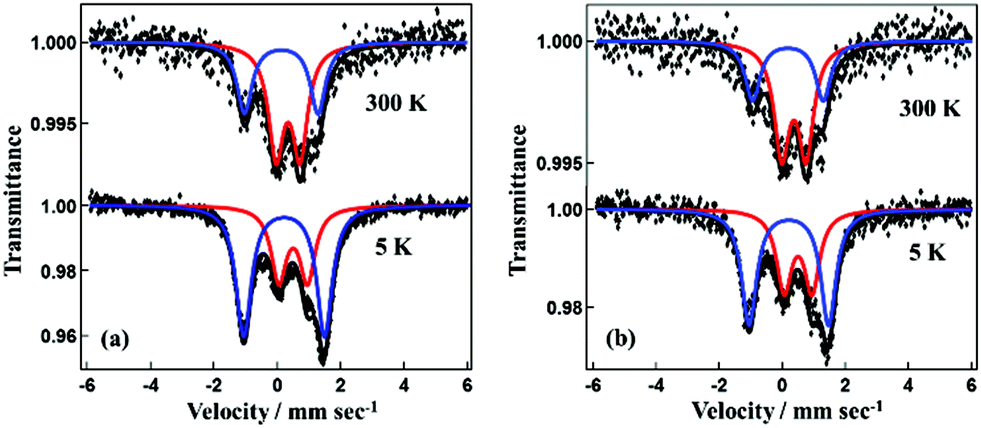 | ||
| Fig. 5 Mössbauer spectra for (a) 1·GO and (b) 1·rGO. | ||
The LIESST experiments for 1·GO and 1·rGO were carried out employing an illuminating semiconductor laser (1000 nm) coupled via an optical fiber to the cavity of a SQUID magnetometer (Fig. 6). The sample was placed on the edge of the optical fiber. On illuminating 1·rGO at 5 K, an increase in its magnetization was observed, suggesting that the photo-induced metastable HS state can be trapped in the 1·rGO hybrid. Relaxation of this metastable state to the ground LS state occurred at about 50 K. For 1·GO, the LIESST effect was not observed. The TEM image of 1·rGO shows the randomly distributed dark spots demonstrating the existence of gathered complexes in which the metal complexes have a shorter intermolecular distance than 1·GO, which revealed that a clustering of the complexes on rGO is a key to produce the LIESST effect of 1·rGO.
 | ||
| Fig. 6 χ g T vs. T plot for 1·GO (red) and 1·rGO (blue). Samples 1·GO (yellow) and 1·rGO (green) were warmed after 1000 nm irradiation at 5 K. | ||
It is worth stating that the coexistence of SCO and electrical conductivity in any graphene hybrid is highly fascinating, as the property coalescence resulted from sustaining the components’ functionality. As SCO cationic species we chose classical Fe3+ complexes of N-donor ligands. The SCO entity [Fe(qsal)2]+ and [Fe(qnal)2]+ become attached and coagulated on the porous GO and rGO framework without suffering any chemical change. The hybrids take the shapes of SCO nanoparticles dispersed on 2D graphene. As chemical reduction results in a coagulated mass of the complex in each case, thermal reduction was employed for the samples to be used for the conductivity measurements. The cations are strongly coupled and arranged closely via π-stacking with rGO matrices. Therefore, the large intramolecular structure transmits the SCO electronic transition quite efficiently within the entire hybrid and signifies the coercive force responsible for the observed SCO behavior. Along with the existence of graphene conductivity the establishment of π-stacking to enhance cooperativity in SCO complexes has been demonstrated to be a successful synthetic strategy for devising GO and rGO based hybrids. The electrons travel along the GO and rGO nanosheets giving rise to the electronic conductivity. Both the conductivity of rGO and SCO behavior of the [Fe(qsal)2]+ and [Fe(qnal)2]+ precursors is maintained solely due to the nature of the physical interaction that occurs between these components in the respective hybrid products. That is, the 2D graphene sheet supports polymeric networking between the SCO entities to yield the observed cooperative effect.
Conclusion
We have observed clear evidence of coupling between the electrical properties of rGO and SCO behavior of metal complexes in new hybrid materials. The graphene based hybrids 1·GO, 2·GO, 1·rGO and 2·rGO were synthesized by employing Coulombic attraction and through the sandwiching of iron(III) complexes within the folded layers of rGO. While the GO hybrids are insulators, the rGO hybrids are conductors. Both the hybrids, 1·rGO and 2·rGO, exhibit electrical conductivity. Furthermore 1·GO and 1·rGO exhibit SCO behavior, and 1·rGO also shows the LIESST effect. We succeeded to control the domain size of metal complexes on GO and rGO. These hybrids belong to the rare category of materials displaying the coexistence of SCO behavior and electrical conductivity. Graphene-based materials with these dual properties are presented here for the first time. We look forward to observing related cooperativity between other SCO entities embedded in GO and rGO nanosheet hybrid materials in the future.Acknowledgements
This work was supported by KAKENHI Grant-in-Aid for Scientific Research (B) 26288026 and Grant-in-Aid for Scientific Research on Innovative Areas 2506 [Science of Atomic Layers].Notes and references
- P. Day, World Scientific, Singapore, 2007 Search PubMed.
- M. Clemente-León, E. Coronado, A. Forment-Aliaga, P. Amorós, J. Ramírez-Castellanos and J. M. González-Calbet, J. Mater. Chem., 2003, 13, 3089 RSC.
- J. M. Lehn, Science, 2002, 295, 2400 CrossRef CAS PubMed.
- E. Coronado, J. R. Galán-Mascarós, C. J. Gómez-García and V. Laukhin, Nature, 2000, 408, 447 CrossRef CAS PubMed.
- E. Ojima, H. Fujiwara, K. Kato and H. Kobayashi, J. Am. Chem. Soc., 1999, 121, 5581 CrossRef CAS.
- E. Coronado, C. Marti-Gastaldo, E. Navarro-Moratalla, A. Ribera, S. J. Blundell and P. J. Baker, Nat. Chem., 2010, 2, 1031 CrossRef CAS PubMed.
- E. Coronado, M. Giménez-Marqués, C. Martí-Gastaldo, G. M. Espallargas, E. Navarro-Moratalla and J. C. Waerenborgh, Inorg. Chem., 2013, 52, 8451 CrossRef CAS PubMed.
- M. R. Karim, H. Takehira, T. Matsui, Y. Murashima, R. Ohtani, M. Nakamura and S. Hayami, Curr. Inorg. Chem., 2014, 4, 191 CrossRef CAS.
- M. R. Karim, H. Shinoda, M. Nakai, K. Hatakeyama, H. Kamihata, T. Matsui, T. Taniguchi, M. Koinuma, K. Kuroiwa, M. Kurmoo, Y. Matsumoto and S. Hayami, Adv. Funct. Mater., 2013, 23, 323 CrossRef CAS PubMed.
- Y. Murashima, R. Ohtani, T. Matsui, H. Takehira, R. Yokota, M. Nakamura, L. F. Lindoy and S. Hayami, Dalton Trans., 2015, 44, 5049 RSC.
- P. Gütlich and H. A. Goodwin, Top. Curr. Chem., 2004, 233, 1 CrossRef.
- W. Vreugdenhil, J. H. Vandiemen, R. A. Degraaf, J. G. Haasnoot, A. M. Vanderkraan, O. Kahn and J. Zarembowitch, Polyhedron, 1990, 9, 2971 CrossRef CAS.
- M. Clemente-León, E. Coronado, M. C. Gimónez-López and F. M. Romero, Inorg. Chem., 2007, 46, 11266 CrossRef PubMed.
- O. Sato, T. Iyoda, A. Fujishima and K. Hashimoto, Science, 1996, 272, 704 CAS.
- V. Niel, J. M. Martínez-Agudo, M. C. Muñoz, A. B. Gaspar and J. A. Real, Inorg. Chem., 2001, 40, 3838 CrossRef CAS.
- J.-F. Létard, P. Guionneau, O. Nguyen, J. S. Costa, S. Marcen, G. Chastanet, M. Marchivie and L. Goux-Capes, Chem. – Eur. J., 2005, 11, 4582 CrossRef PubMed.
- W. Kosaka, K. Nomura, K. Hashimoto and S. Ohkoshi, J. Am. Chem. Soc., 2005, 127, 8590 CrossRef CAS PubMed.
- A. B. Gaspar, V. Ksenofontov, M. Seredyuk and P. Gütlich, Coord. Chem. Rev., 2005, 249, 2661 CrossRef CAS PubMed.
- V. Niel, A. L. Thompson, M. C. Muñoz, A. Galet, A. E. Goeta and J. A. Real, Angew. Chem., Int. Ed., 2003, 42, 3760 CrossRef CAS PubMed.
- M. Clemente-León, E. Coronado, M. López-Jordà, J. C. Waerenborgh, C. Desplanches, H. Wang, J.-F. Létard, A. Hauser and A. Tissot, J. Am. Chem. Soc., 2013, 135, 8655 CrossRef PubMed.
- M. Clemente-León, E. Coronado and M. López-Jordà, Dalton Trans., 2010, 39, 4903 RSC.
- H. Akamatu, H. Inokuchi and Y. Matsunaga, Nature, 1954, 173, 168 CrossRef PubMed.
- E. Coronado and P. Day, Chem. Rev., 2004, 104, 5419 CrossRef CAS PubMed.
- L. Ouahab, Coord. Chem. Rev., 1998, 178–180, 1501 CrossRef CAS.
- E. Coronado and J. R. Galan-Mascarós, J. Mater. Chem., 2005, 15, 66 RSC.
- K. Takahashi, H.-B. Cui, Y. Okano, H. Kobayashi, H. Mori, H. Tajima, Y. Einaga and O. Sato, J. Am. Chem. Soc., 2008, 130, 6688 CrossRef CAS PubMed.
- B. Djukic and M. T. Lemaire, Inorg. Chem., 2009, 48, 10489 CrossRef CAS PubMed.
- B. Djukic, T. Seda, S. I. Gorelsky, A. J. Lough and M. T. Lemaire, Inorg. Chem., 2011, 50, 7334 CrossRef CAS PubMed.
- S. Dorbes, L. Valade, J. A. Real and C. Faulmann, Chem. Commun., 2005, 69 RSC.
- K. Takahashi, H.-B. Cui, Y. Okano, H. Kobayashi, Y. Einaga and O. Sato, Inorg. Chem., 2006, 45, 5739 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS PubMed.
- T. Shimizu, Y. Komatsu, H. Kamihata, Y. H. Lee, A. Fuyuhiro, S. Iijima and S. Hayami, J. Inclusion Phenom. Macrocyclic Chem., 2011, 71, 363 CrossRef CAS.
- S. Hayami, Z. Gu, H. Yoshiki, A. Fujishima and O. Sato, J. Am. Chem. Soc., 2001, 123, 11644 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906 CrossRef CAS PubMed.
- M. R. Karim, K. Hatakeyama, T. Matsui, H. Takehira, T. Taniguchi, M. Koinuma, Y. Matsumoto, T. Akutagawa, T. Nakamura, S.-I. Noro, T. Yamada, H. Kitagawa and S. Hayami, J. Am. Chem. Soc., 2013, 135, 8097 CrossRef CAS PubMed.
- S. Hayami, K. Hiki, T. Kawahara, Y. Maeda, D. Urakami, K. Inoue, M. Ohama, S. Kawata and O. Sato, Chem. – Eur. J., 2009, 15, 3497 CrossRef CAS PubMed.
- P. Cui, J. Lee, E. Hwang and H. Lee, Chem. Commun., 2011, 47, 12370 RSC.
- F. Tuinstra and J. L. Koenig, J. Chem. Phys., 1970, 53, 1126 CrossRef CAS PubMed.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter, 2000, 61, 14095 CrossRef CAS.
- M. C. Hsiao, S. H. Liao, M. Y. Yen, P. I. Liu, N. W. Pu, C. A. Wang and C. C. Ma, Appl. Mater. Interfaces, 2010, 2, 3092 CrossRef CAS PubMed.
- Y. Ikeda, M. R. Karim, H. Takehira, K. Hatakeyama, T. Matsui, T. Taniguchi, M. Koinuma, Y. Matsumoto and S. Hayami, Chem. Lett., 2014, 43, 486 CrossRef CAS.
- Z. Zafar, Z. H. Ni, X. Wu, Z. X. Shi, H. Y. Nan, J. Bai and L. T. Sun, Carbon, 2013, 61, 57 CrossRef CAS PubMed.
- R. Podila, J. Chacón-Torres, J. T. Spear, T. Pichler and P. Ayala, Appl. Phys. Lett., 2012, 101, 123108 CrossRef PubMed.
- M. R. Karim, Y. Ikeda, T. Ide, S. Sugimoto, K. Toda, Y. Kitamura, T. Ihara, T. Matsui, T. Taniguchi, M. Koinuma, Y. Matsumoto and S. Hayami, New J. Chem., 2014, 38, 2120 RSC.
- T. Szabó, O. Berkesi, P. Forgó, K. Josepovits, Y. Sanakis, D. Petridis and I. Dékány, Chem. Mater., 2006, 18, 2740 CrossRef.
- D. W. Boukhvalov and M. I. Katsnelson, J. Am. Chem. Soc., 2008, 130, 10697 CrossRef CAS PubMed.
- Y. Matsumoto, M. Koinuma, S. Y. Kim, Y. Watanabe, T. Taniguchi, K. Hatakeyama, H. Tateishi and S. Ida, Appl. Mat. Int., 2010, 2, 3461 CrossRef CAS PubMed.
- D. Chen, H. Feng and J. Li, Chem. Rev., 2012, 112, 6027 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qi00097a |
| This journal is © the Partner Organisations 2015 |