DOI:
10.1039/C5QI00054H
(Research Article)
Inorg. Chem. Front., 2015,
2, 671-677
Stable iso-bacteriochlorin mimics from porpholactone: effect of a β-oxazolone moiety on the frontier π-molecular orbitals†
Received
31st March 2015
, Accepted 12th June 2015
First published on 12th June 2015
Abstract
Iso-bacteriochlorins known as siroheme in some reductases, featured with two adjacent reduced pyrrole rings, have distinctive electronic structures from porphyrin, chlorin and bacteriochlorin analogues. However, the synthesis of such cofactor mimics from hydrogenation of chlorin or porphyrin is associated with drawbacks of uncertain regioselectivity and stability. In this work, we present the first example of selective hydrogenation of the adjacent pyrroles in porphyrin or porpholactone free bases assisted by the Woollins reagent (WR). More importantly, adjacent-dihydroporpholactone (1a) displays iso-bacteriochlorin type spectral features and much higher stability under oxidative conditions, compared to the tetrahydroporphyrin analogue (2a). Analysis of magnetic circular dichroism (MCD) spectra and DFT calculations for the frontier π-molecular orbitals for 1a and 2a reveals the significant effect of a β-oxazolone moiety replacement on lowering the HOMO energy level and enhancing the stability resistant to oxidative conditions.
Introduction
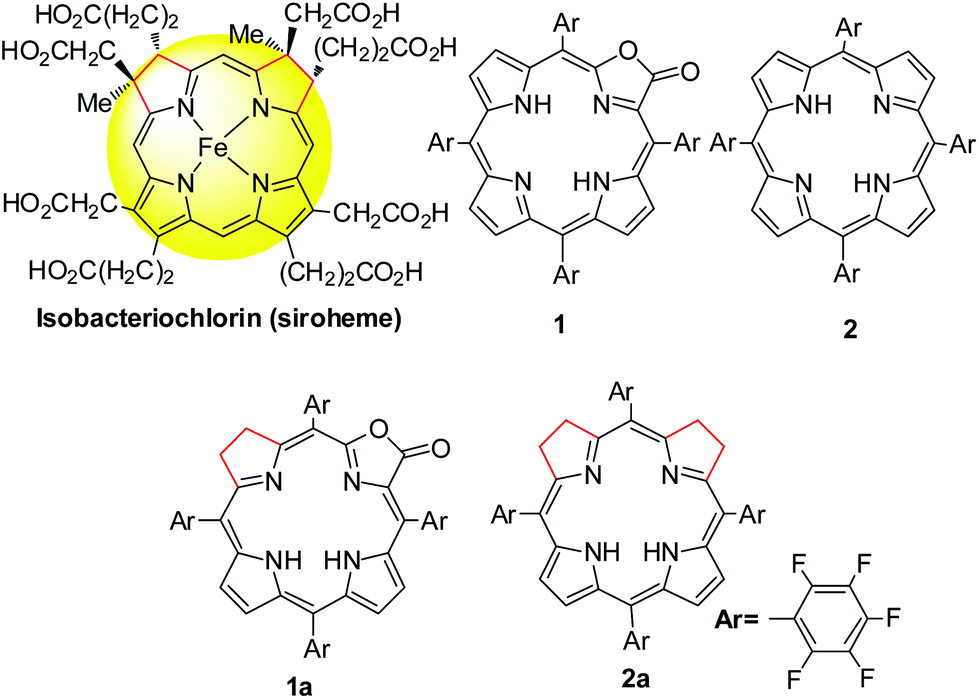
Tetrapyrroles are distributed as biological cofactors with diverse biochemical roles such as light absorption characteristics, redox catalytic reactivity, etc.1 Significant diversity in biological functions suggests the importance of electronic differences, arising from the saturation levels of tetrapyrrole rings with their broad structural similarity.2 To decipher the chemical basis, the development of synthetic hydroporphyrins to mimic the electronic structures of natural tetrapyrroles becomes one of the key areas in current synthetic porphyrin chemistry.3 An iso-bacteriochlorin, featured with two adjacent reduced pyrrole rings, plays an important role in sulfite and nitrite reductases as a prosthetic group4 (known as siroheme, Scheme 1) and as a key intermediate in the biosynthetic pathway to vitamin B12.5 However, synthesis of such mimics is challenging and fraught with a number of problems such as (1) stability, suffering from an easy oxidation back to a porphyrin (or other products); and (2) uncertain regioselectivity, lack of methodology to selective hydrogenation of adjacent pyrroles from chlorin or porphyrin free bases. Although total synthesis or semi-synthesis by the joining of eastern and western dipyrrolic components have been reported,6 multiple synthetic steps with small scales thus diminish the practical value. Thus, a new approach to stable iso-bacteriochlorin mimics is highly desirable to expand the scope of hydroporphyrins,2a,7 in a manner that should facilitate the fundamental understanding of the electronic structures and further applications.
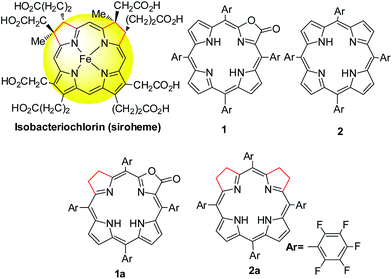 |
| | Scheme 1 Structural formulae of siroheme, porphyrin and porpholactone and hydrogenated analogues. | |
As our continued interest in porpholactone,8 in which one pyrrole of porphyrin is replaced by an oxazolone (or lactone) moiety (Scheme 1), we envisioned that this replacement might facilitate to stabilize the hydroporphyrins by lowering the HOMO energy level. Moreover, for the partial saturated oxazolone replacement, selective hydrogenation of the adjacent pyrroles of the oxazolone moiety might achieve iso-bacteriochlorin mimics. This would circumvent the regioselectivity issue by tuning the electronic effect of substituents on the porphyrin periphery, according to a theoretical study by Bruhn and Brückner.9 In this work, we report the first example of selective hydrogenation of the adjacent pyrroles in porphyrin or porpholactone free bases assisted by the Woollins reagent (PhPSe2)2. More importantly, adjacent-dihydroporpholactone (1a) displays iso-bacteriochlorin like spectral features and much higher stability under oxidative conditions, compared to tetrahydroporphyrin analogue (2a). To decipher the effect of β-oxazolone on the electronic structures, we performed magnetic circular dichroism (MCD) spectroscopy and DFT calculations to analyze the relative energies of the frontier π-molecular orbitals and hence on the optical properties. These results suggested that the higher stability of 1a is due to the decreased HOMO energy level after replacing the β-oxazolone moiety. Furthermore, we applied 1a as a cell imaging agent by encapsulating into poly(lactide-co-glycolide) nanoparticles and demonstrated its good stability and luminescence. Thus, this work provides an access to stable iso-bacteriochlorin like analogue and highlights the importance of β-oxazolone moiety replacement on further studying natural tetrapyrrole mimics.
Results and discussion
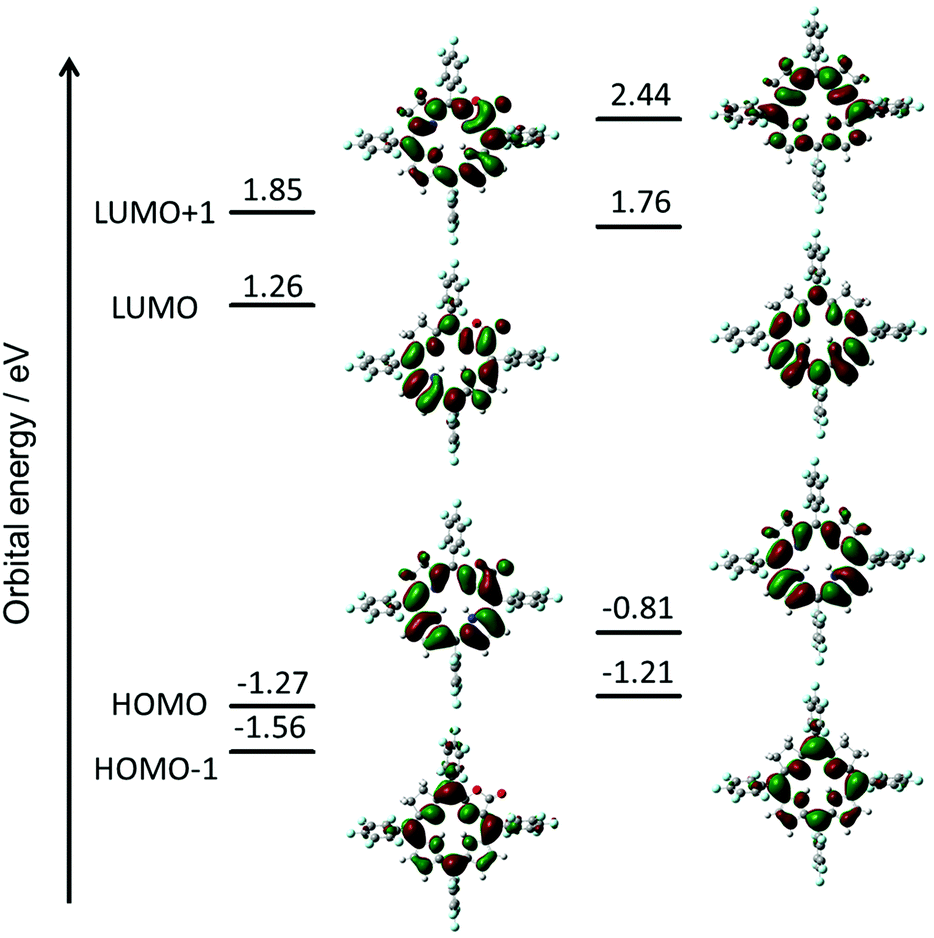
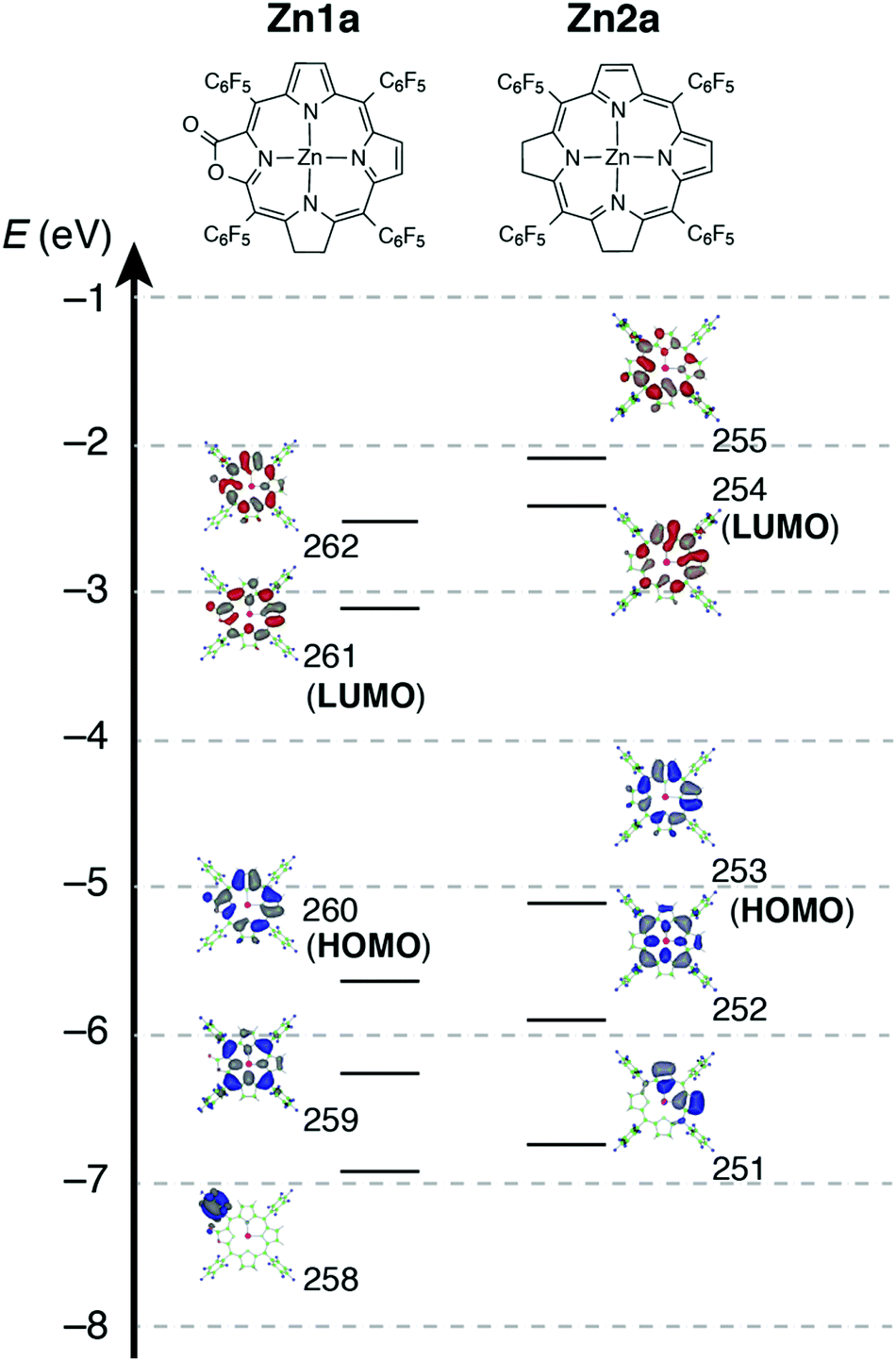
To evaluate the effect of the β-oxazolone moiety on the stability of hydroporphyrins, we firstly estimated the molecular structures and molecular orbital (MO) diagrams of adjacent-dihydro-porpholactones (1a) and tetrahydroporphyrin analogue (2a) derivatized from tetrapentafluorophenylporpholactone (H2F20TPPL, 1) and tetrapentafluorophenylporphyrin (H2F20TPP, 2), at the B3LYP/6-31G* level using the Gaussian 09 package, on the basis of Gourterman's four orbital model. As shown in Fig. 1, for 1a and 2a, the hydrogenation of tetrapyrrole rings leads to less degenerate MOs, which is in accordance with those for previously reported iso-bacteriochlorin analogues.2c For the β-oxazolone moiety that participated in the π-conjugation, the energies of four frontier orbitals of 1a are evidently lowered than those of 2a, especially for HOMO and LUMO levels (ca. 0.46 and 0.50 eV, respectively). Thus, on the basis of DFT calculations, the replacement of the β-oxazolone moiety renders iso-bacteriochlorin mimics 1a more resistant to oxidative conditions.
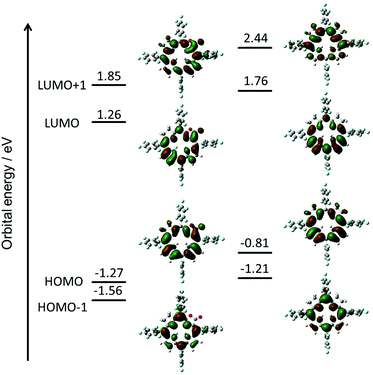 |
| | Fig. 1 Molecular orbital diagrams of 1a (left) and 2a (right), optimized at the B3LYP/6-31 G* level using the Gaussian 09 software package. | |
Synthesis of iso-bacteriochlorin mimics 1a and 2a
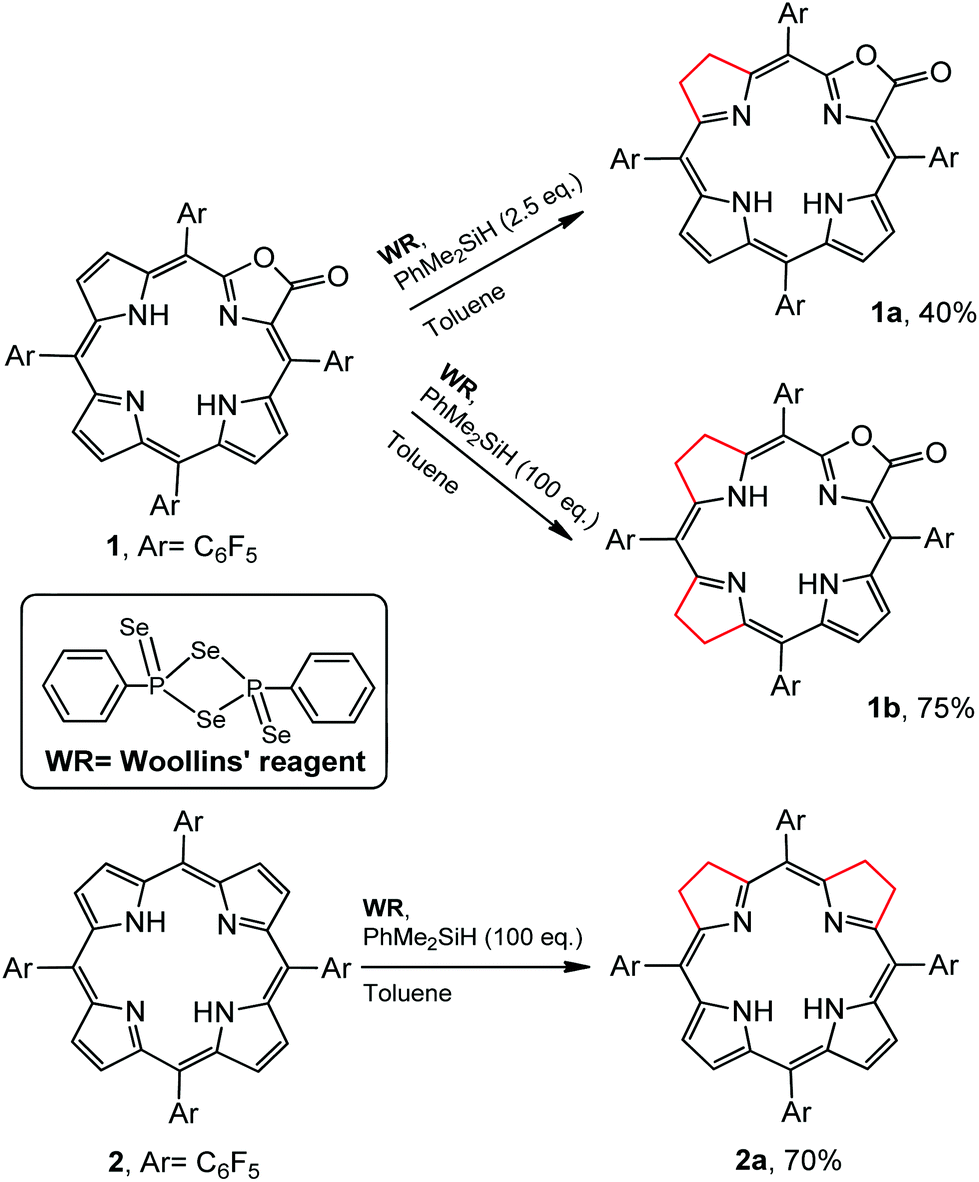
Regioselectivity is another important issue yet to be addressed in the synthesis of iso-bacteriochlorin mimics. Generally, the reactions of diimide based reductants,7j,10 or electrophilic agents such as OsO4,8h,11 ozone12 or dienes13 or 1,3-dipoles such as azomethine ylides, nitrones, or nitrile oxides,14 with chlorin or porphyrin free bases afforded bacteriochlorin type hydroporphyrin (the opposite pyrroles were hydrogenated), while with metallochlorin or porphyrin lead to metalloiso-bacteriochlorins. In this work, attempts to hydrogenation of 1 to 1a using previous methods such as photoreduction7i or reducing with diimide7j failed. We then turned our attention to the selenium-assisted reduction system based on Woollins’ reagent (WR).15 Since Jaisankar et al. demonstrated the effectiveness in hydrogenation of aromatic ketones,16 WR based reduction procedures had been extended to selective hydrogenation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in α,β-unsaturated carbonyl compounds, though the mechanisms were still unclear.17 As shown in Scheme 2, the reaction of 1 eq. WR, 1 and 4 eq. PhMe2SiH in refluxed toluene gave the adjacent-dihydroporpholactone 1a in an isolated yield of 40%. We did not observe the product that Se replaces an O atom of the carbonyl group. ESI-MS showed a molecular ion peak at m/z = 995.0543 (ca. 995.0557), consistent with the dihydrogenation of one pyrrole in 1a. The 1H NMR spectrum (CDCl3) displayed three sets of peaks at 7.28, 7.54 and 7.74 ppm (β-H, 4H), two sets of multiple peaks at 3.86 and 3.82 ppm (C–H at reduced pyrrole, 4H) and two broad protons at 3.97 and 4.86 ppm (N–H, 2H) which would disappear after coordination with the Zn2+ ion (ESI†). The splitting of 19F signals at −138, −153 and −162 ppm illustrated lower symmetry than that of porpholactone 1. The vibration of CO at 1780 cm−1 on the IR spectrum showed that the lactone moiety remained intact (1766 cm−1 for 1). The UV-vis spectrum of 1a (will discuss in the next section) displays iso-bacteriochlorin type absorption. Similarly, tetrahydroporphyrin 2a was obtained using 2 as a precursor in the isolated yield of 70% (ESI†). A large excess amount of silane (100 equiv.) in the synthesis of 2a was used, probably due to two adjacent pyrroles that required to be hydrogenated other than one pyrrole in 1a. To confirm this, we increased the amount of silane to 100 equiv. in the reaction of 1 with WR, and found that tetrahydroporpholactone 1b was obtained in the yield of 75% (characterization in the ESI†). 1b and 2a also exhibit iso-bacteriochlorin type spectra as previously reported tetraphenylporphyrin analogues.2a,18 It is worthy to note that, in the absence of WR or PhMe2SiH, the formation of 1a, 1b or 2a was not observed. Interestingly, no reaction was observed using metalloporpholactone Zn1 or metalloporphyrin Zn2 as a starting material under the same conditions. Metalation of 1a, 1b and 2a with Zn(OAc)2 in methanol produced Zn1a, Zn1b and Zn2a (the details listed in the ESI†). These results demonstrated that the protocol containing WR and silane is effective to directly reduce the adjacent double bond of porpholactone and porphyrin free bases.
C bond in α,β-unsaturated carbonyl compounds, though the mechanisms were still unclear.17 As shown in Scheme 2, the reaction of 1 eq. WR, 1 and 4 eq. PhMe2SiH in refluxed toluene gave the adjacent-dihydroporpholactone 1a in an isolated yield of 40%. We did not observe the product that Se replaces an O atom of the carbonyl group. ESI-MS showed a molecular ion peak at m/z = 995.0543 (ca. 995.0557), consistent with the dihydrogenation of one pyrrole in 1a. The 1H NMR spectrum (CDCl3) displayed three sets of peaks at 7.28, 7.54 and 7.74 ppm (β-H, 4H), two sets of multiple peaks at 3.86 and 3.82 ppm (C–H at reduced pyrrole, 4H) and two broad protons at 3.97 and 4.86 ppm (N–H, 2H) which would disappear after coordination with the Zn2+ ion (ESI†). The splitting of 19F signals at −138, −153 and −162 ppm illustrated lower symmetry than that of porpholactone 1. The vibration of CO at 1780 cm−1 on the IR spectrum showed that the lactone moiety remained intact (1766 cm−1 for 1). The UV-vis spectrum of 1a (will discuss in the next section) displays iso-bacteriochlorin type absorption. Similarly, tetrahydroporphyrin 2a was obtained using 2 as a precursor in the isolated yield of 70% (ESI†). A large excess amount of silane (100 equiv.) in the synthesis of 2a was used, probably due to two adjacent pyrroles that required to be hydrogenated other than one pyrrole in 1a. To confirm this, we increased the amount of silane to 100 equiv. in the reaction of 1 with WR, and found that tetrahydroporpholactone 1b was obtained in the yield of 75% (characterization in the ESI†). 1b and 2a also exhibit iso-bacteriochlorin type spectra as previously reported tetraphenylporphyrin analogues.2a,18 It is worthy to note that, in the absence of WR or PhMe2SiH, the formation of 1a, 1b or 2a was not observed. Interestingly, no reaction was observed using metalloporpholactone Zn1 or metalloporphyrin Zn2 as a starting material under the same conditions. Metalation of 1a, 1b and 2a with Zn(OAc)2 in methanol produced Zn1a, Zn1b and Zn2a (the details listed in the ESI†). These results demonstrated that the protocol containing WR and silane is effective to directly reduce the adjacent double bond of porpholactone and porphyrin free bases.
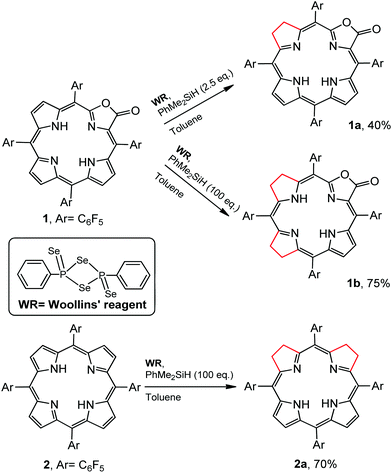 |
| | Scheme 2 Synthetic routes for 1a, 1b and 2a. | |
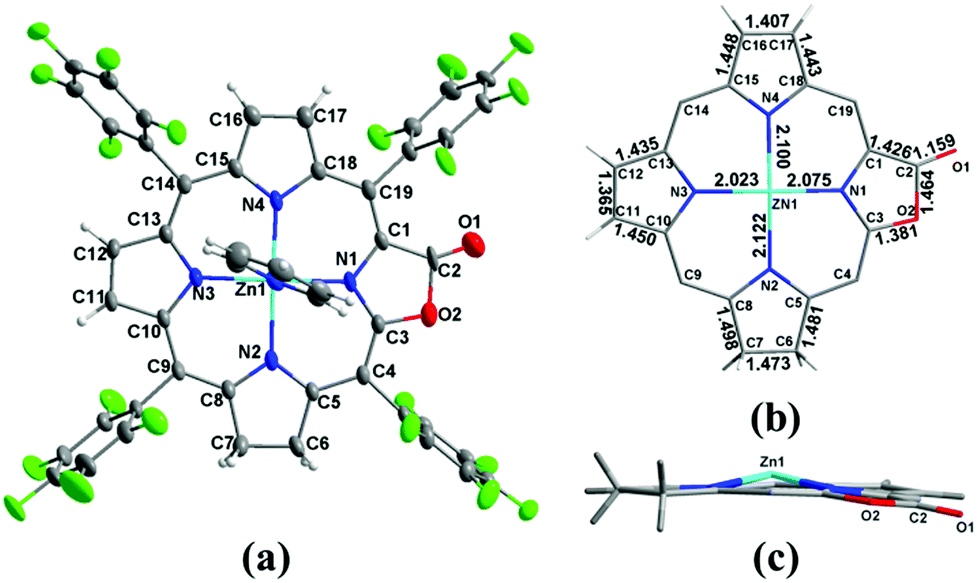
To clarify where hydrogenation takes place, we tried to grow the single crystals of 1a however, we could not get the plausible data due to the disorder of the lactone moiety. Metalation of 1a with the Zn2+ ion produced Zn1a and the single crystal suitable for X-ray diffraction could be obtained in the presence of pyridine (CCDC: 1047515). In Fig. 2, the ORTEP structure shows that the additional hydrogens are located on the pyrrole adjacent to the oxazolone ring (oxa-atom side). It is reflected by the elongated C6–C7 bond length (1.473 Å) relative to the other pyrroles (C11–C12 and C16–C17: 1.365 and 1.407 Å), which is comparable to 1.479 and 1.497 Å in previously reported adjacent-(tetrahydrotetraphenylporphinato)(pyridine)zinc(II).19 Similar 1.481 Å and 1.498 Å are also observed in C5–C6 and C7–C8 bond distances (1.472 Å to 1.508 Å in the case of (adjacent-tetrahydrotetraphenylporphinato)(pyridine)zinc(II)). The Zn–N2 distance is 2.122 (5) Å, which is longer than the other three Zn–N distances (2.023 (4) Å, 2.075 (4) Å and 2.100 (5) Å). The torsion angle of CαCβCβCα of the reduced pyrrole ring is 4.23°, indicating the twist (0.86° and 2.40° for the other two conjugated pyrrole rings) in the porphyrin ring.
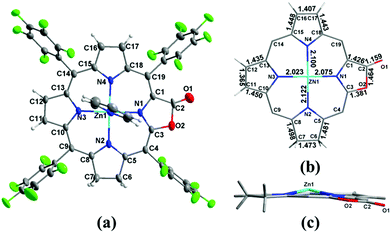 |
| | Fig. 2 (a) ORTEP diagram of Zn1a (thermal ellipsoids at the 30% probability level); (b) selected bond distances (Å) for the core of Zn1a; (c) edge-on-views of the core of Zn1a along the N2–N3–N4 planes. | |
Photophysical properties
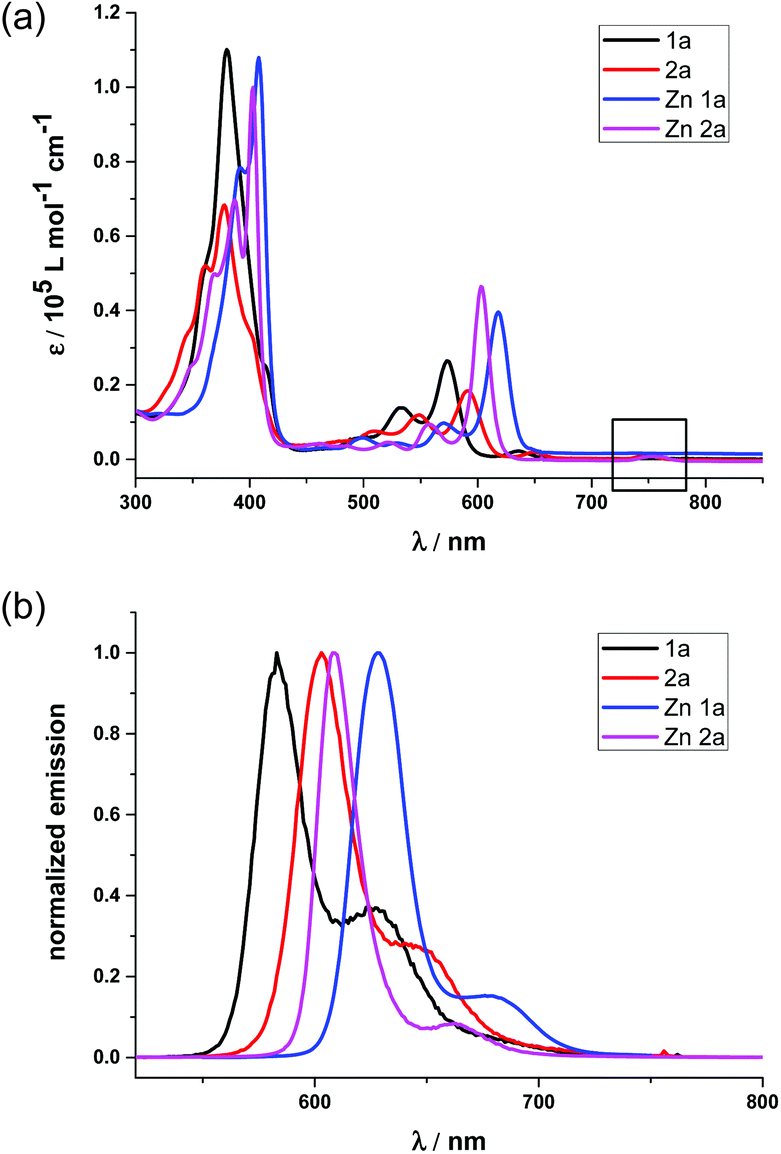
To better demonstrate the effect of the replacement of the β-oxazolone moiety, we discuss the photophysical properties of 1a and 2a in the context. The electronic absorption of 1a, 2a and their zinc complexes in CH2Cl2, as shown in Fig. 3(a), display iso-bacteriochlorin type spectra with broader, split, and blue-shifted Soret bands.2d,4f,18a Compared with 1 and 2, 1a and 2a also exhibit broad, intense and blue-shifted Q bands at 500–610 nm, indicating the degeneracy of molecular orbitals and slightly increasing HOMO–LUMO gaps due to lower molecular symmetry after hydrogenation. For 1a, there is small absorption with maxima at 636 nm, which disappears after metallation with the Zn2+ ion (Fig. 3(a)). Thus, this absorption is assumed to be a partial protonation of pyrroles, further verified by using the acid titration experiment as shown in Fig S7.† However, for 2a, the absorption at ca. 750 nm does not disappear even in Zn2a, which might be due to the contamination of trace bacteriochlorin for less selective reduction of porphyrin 2. Compared with the free bases, Zn complexes show red shifts of the absorption bands. In particular, the difference of Q(0,0) bands between 1a and Zn1a is 43 nm, which is larger than that between 2a and Zn2a (14 nm), respectively. The red shifts of absorption bands indicate less macrocycle distortion of Zn complexes and enhancing π-conjugation.
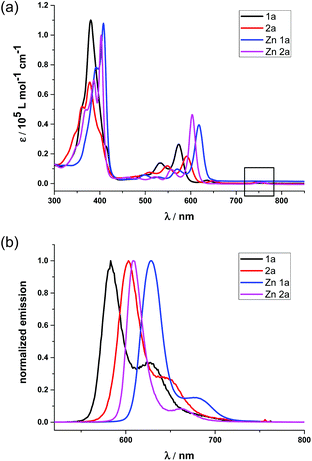 |
| | Fig. 3 (a) Absorption spectra of 1a (black), 2a (red), Zn1a (blue) and Zn2a (purple) in CH2Cl2; (b) normalized emission spectra of 1a (black), 2a (red), Zn1a (blue) and Zn2a (purple) in CH2Cl2. Concentrations are all 5.0 × 10−6 M, and all excited at maximum absorption. | |
The fluorescence spectra of 1a and 2a in Fig. 3(b) display peak maxima (λmax) at 583 nm and 603 nm with a shoulder at 625 and 654 nm, respectively. The fluorescence quantum yields and fluorescence lifetimes were determined to be 0.47 and 3.88 ns for 1a, 0.55 and 5.63 ns for 2a, respectively. After metallization, the quantum yield and lifetime of Zn1a were 0.13 and 0.70 ns, while 0.08 and 1.08 ns for Zn2a. Thus, radiative and non-radiative decay rates were calculated to be kr = 1.2 × 108 and knr = 1.4 × 108 s−1 for 1a, kr = 9.8 × 107 and knr = 9.8 × 107 s−1 for 2a, kr = 1.9 × 108 and knr = 1.2 × 109 s−1 for Zn1a, kr = 7.4 × 107 and knr = 8.5 × 108 s−1 for Zn2a, respectively.
The electrochemical properties were studied by cyclic voltammetry in CH2Cl2 (vs. ferrocene 0.45 V as a standard, Table S2, Fig. S13 and S14†). Compared with 1 and 2, 1a and 2a display cathodic shifts of ca. 0.37 and 0.61 V, respectively, for the first oxidation potentials; and ca. 0.21–0.33 V for the first reduction potentials. The first oxidation potential of 1a (1.33 V) is more positive than 2a (0.92 V) whereas there is a lesser difference in the first reduction potentials between 1a (−0.78 V) and 2a (−1.13 V). A similar trend was observed for Zn1a and Zn2a, in which the first oxidation potential of Zn1a (0.99 V) is 0.37 V higher than that of Zn2a (0.62 V). Again, these electrochemical studies clearly showed that the replacement of the β-oxazolone moiety lowers the first oxidative potentials of hydroporphyrins more than the first reduction potentials, given that similar HOMO−LUMO gaps are obtained from UV-vis absorption for 1a and 2a, Zn1a and Zn2a. This is consistent with the trend of energies of four frontier orbitals and HOMO−LUMO gaps for 1a and 2a based on DFT calculations.
Stability
Stabilities of 1a, 2a, Zn1a and Zn2a were examined under oxidative conditions using m-CPBA, DDQ and light irradiation (ESI†). We used UV-vis absorption and 1H NMR spectroscopy to monitor the reaction process. 1a and Zn1a exhibited high stability and remained intact when treated with 10 eq. m-CPBA for 1 day or irradiated with light at 365 nm for 2 h. 2a decomposed upon addition of m-CPBA in 2 h or light irradiation for 90 min (88%, calculated based on 1H NMR integration). Zn2a was very unstable toward oxidation or UV irradiation, and even decomposition was observed on the silica gel column. These results clearly demonstrated that the oxazolone replacement indeed stabilized the reduced porphyrinoid structures, in line with electrochemical and theoretical studies.
MCD spectroscopy and DFT calculation
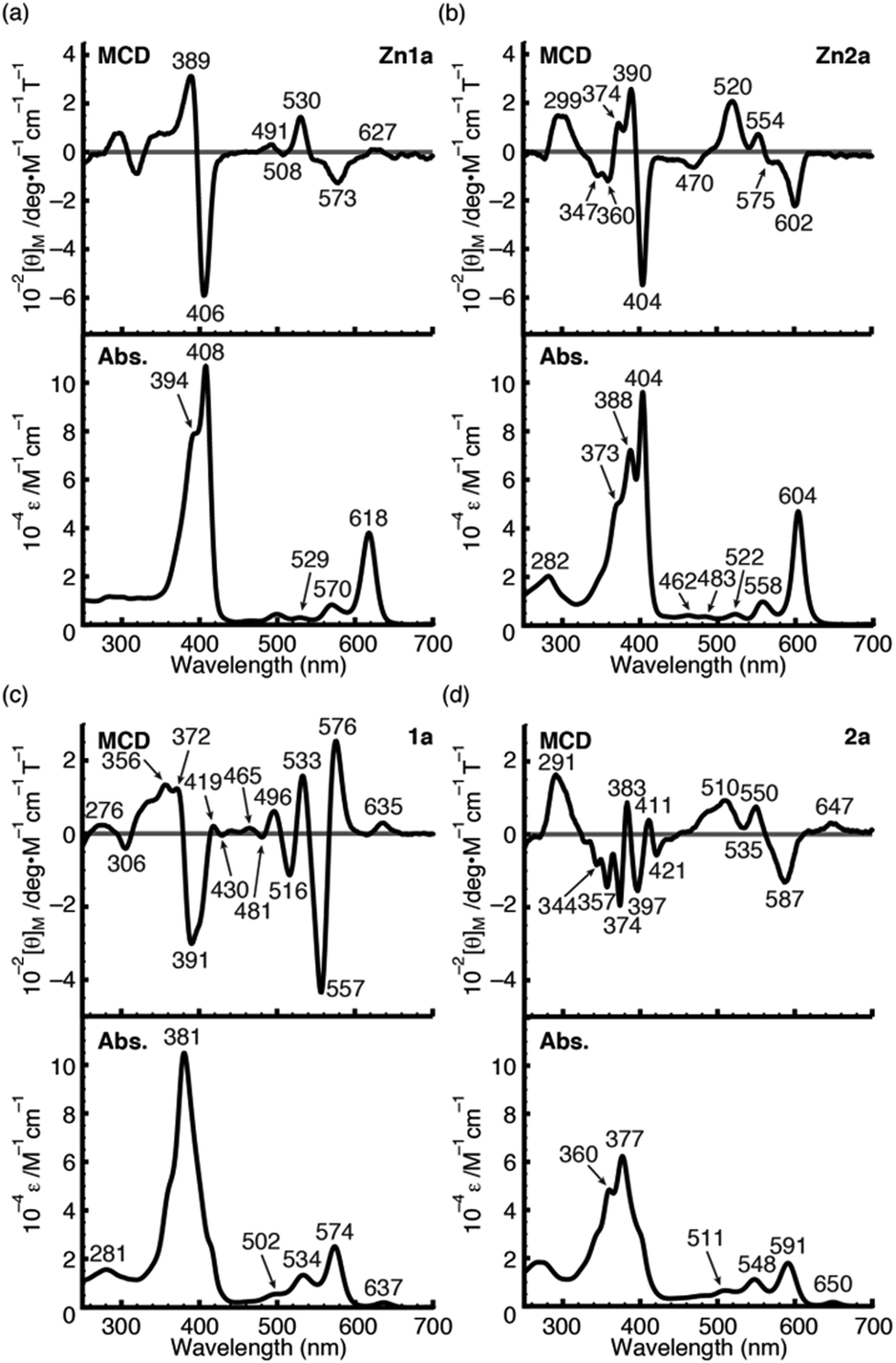
To further demonstrate the advantage of porpholactone, we used 1a, Zn1a, 2a and Zn2a to discuss the effect of oxazolone replacement on the photophysical properties and electronic structures. The electronic absorption and MCD spectra of these complexes in CH2Cl2 are shown in Fig. 4. Compared with normal porphyrins which have four unsaturated pyrrole rings, the intensity of the Q band is much stronger, so that the Q/Soret intensity ratio is stronger than in normal porphyrins. According to the Gouterman's four orbital theory that has been applied in understanding spectra of porphyrinoids,20 this indicates that the energy difference between the HOMO and HOMO−1 (ΔHOMO) is larger than that in normal porphyrins. Due to the uncertainty of the position of two pyrrole protons and the presence of several isomers, the spectra of metal-free 1a and 2a are more complex than those of the Zn1a and Zn2a. For 1a, there is a small absorption with a maximum at 636 nm. This absorption is ascribed to the partial protonation of pyrroles, which is verified by using the acid titration experiment, and which disappears after metallation with the Zn2+ ion. The Zn complexes show red-shifted absorption compared with those of metal-free species. In particular, the difference of the Q00 bands between 1a and Zn1a is 43 nm, which is larger than that between 2a and Zn2a (14 nm). The spectra of 1a and Zn1a are broadly similar to those of iso-bacteriochlorin whose two pyrrole rings at adjacent positions are reduced.18a,21 The splitting of the Q band is theoretically not large, different from that of bacteriochlorin which has saturated pyrrole rings at opposite positions.22
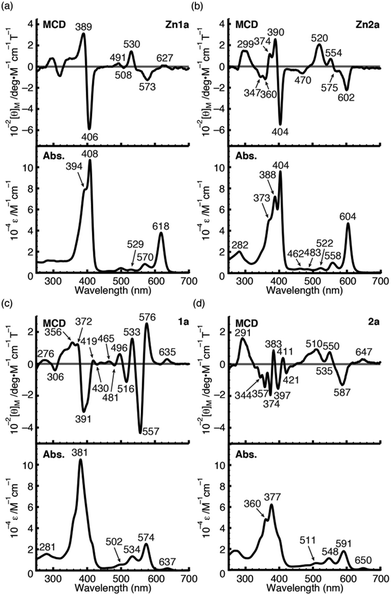 |
| | Fig. 4 MCD (top) and UV/Vis (bottom) absorption spectra of Zn1a (a), Zn2a (b), 1a (c) and 2a (d) in CH2Cl2. | |
For complexes in this study, the MCD spectra are all contribution of Faraday B terms.23 The observed spectra were interpreted in consideration of the results of MO calculations. Although the MCD spectra are complex for metal-free species, the most decisive difference between 1a and 2a is that the MCD sign for the Q00 band is positive for 1a and negative for 2a in ascending energy, experimentally suggesting that the ΔHOMO is smaller than ΔLUMO (the energy difference between the LUMO and LUMO+1) for 1a and opposite for 2a.23 The MCD spectra of Zn1a and Zn2a appear similar at a glance, but there is a decisive difference in the Q band region, namely, for Zn2a, a clear negative MCD envelope was observed corresponding to the Q00 absorption peak at 604 nm, indicating that ΔHOMO is obviously larger than ΔLUMO.23 Meanwhile for Zn1a, no (or very small positive) MCD intensity was observed associated with the Q00 band at 616 nm, indicating experimentally that ΔHOMO is nearly equal to ΔLUMO. Judging from the MCD sign, another important information is that the peak at 570 nm for Zn1a and 558 nm for Zn2a correspond to the split components of the Q band, since the interacting MCD B-terms give signals of an opposite sign.23,24 Taking all the MCD data of 1a, 2a, Zn1a, and Zn2a into account, it is experimentally concluded that ΔHOMO is much larger than ΔLUMO for 2a and Zn2a, while that the ΔHOMO is similar to or very slightly smaller than ΔLUMO for 1a and Zn1a. We have calculated molecular orbitals (MOs) of Zn1a and Zn2a, hence we don't need to consider the position of pyrrole protons that occurred for metal-free 1a and 2a. The data are shown in Fig. 5. Compared with the MOs of Zn2a, those of Zn1a are all from the calculation, and the above relationship on the size of ΔHOMO and ΔLUMO was nicely reproduced, i.e. they are 0.61 and 0.60 eV for Zn1a and 0.79 and 0.32 eV for Zn2a, respectively.
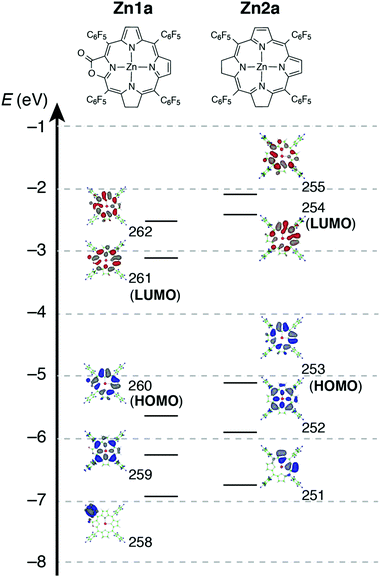 |
| | Fig. 5 Calculated frontier orbitals of Zn1a (left) and Zn2a (right). | |
Fluorescence and cell imaging
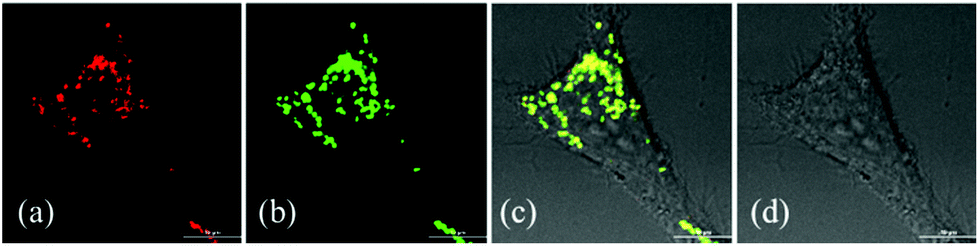
Since β-lactonization of the pyrrole ring renders hydroporphyrins higher stability, we used 1a as an example to demonstrate their potential application in cellular imaging. To make it water-soluble, we used the modified solvent extraction/evaporation single-emulsion method to prepare 1a-loaded poly(lactide-co-glycolide) nanoparticles (PLGA NPs) as reported.25 The intracellular luminescence and subcellular distribution of 1a-NPs were investigated using the LysoTracker® Green DND-26 by confocal laser scanning microscopy (CLSM). HeLa cells were co-incubated with 1a-NPs with a final concentration of 10 μM in complete culture medium for 24 h at 37 °C. As shown in Fig. 6, 1a-NPs possess good cell membrane permeability and leads to the perinuclear punctate red fluorescence in HeLa cells. It mainly distributes in lysosomal/endosomal compartments and exhibits a co-localization level of approximately 0.92 with the LysoTracker® Green DND-26. Thus, preliminary imaging results show 1a-NPs can be internalized into living cells and detected by confocal imaging, which offers the prerequisite and convenience for further biological applications.
 |
| | Fig. 6 Co-localization of 1a-NPs (10 μM) with the LysoTracker® Green DND-26 in HeLa cells. (a) Fluorescence image of 1a-NPs; (b) fluorescence image of the LysoTracker® Green DND-26; (c) the merge of (a), (b) and (d); (d) differential interference contrast (DIC) image. Scale bar: 10 μm. | |
Conclusions
Taken together, we developed a new approach based on the Woollins reagent to prepare iso-bacteriochlorin mimics from porpholactone or porphyrin free bases. More importantly, with an oxazolone moiety replacement, β-adjacent dihydroporpholactone (1a) exhibits high stability toward oxidative conditions and photo-irradiation. A detailed analysis of optical spectra and TD-DFT calculations reveal that 1a and 2a possess electronic structures similar to iso-bacteriochlorins and the significant effect of the oxazolone moiety on lowering the HOMO energy level. Furthermore, cellular uptake and subcellular localization investigations have demonstrated the potential utility of these compounds in the biological studies. The application of such iso-bacteriochlorin analogues as ligands to mimic the reactivity of sulfite and nitrite reductases is currently under investigation.
Acknowledgements
This project was supported by the National Scientific Foundation of China (grants no. 21271013, 21321001) and the National Key Basic Research Support Foundation of China (NKBRSFC) (2013CB933402, 2015CB856300).
Notes and references
-
(a)
T. D. Lash, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, 2000, vol. 2, pp. 125–200 Search PubMed;
(b) J. Mack, M. J. Stillman and N. Kobayashi, Coord. Chem. Rev., 2007, 251, 429–453 CrossRef CAS PubMed;
(c)
J. L. Sessler, A. Gebauer and S. J. Weghorn, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, 2000, vol. 2, pp. 55–124 Search PubMed;
(d) H. J. Xu, J. Mack, A. B. Descalzo, Z. Shen, N. Kobayashi, X. Z. You and K. Rurack, Chem. – Eur. J., 2011, 17, 8965–8983 CrossRef CAS PubMed.
-
(a) J. A. Cavaleiro, M. G. Neves, A. C. Tome, A. Silva, M. A. Faustino, P. S. Lacerda and A. M. Silva, J. Heterocycl. Chem., 2000, 37, 527–534 CrossRef CAS PubMed;
(b) A. Ghosh, J. Phys. Chem. B, 1997, 101, 3290–3297 CrossRef CAS;
(c) N. Otero, S. Fias, S. Radenković, P. Bultinck, A. M. Graña and M. Mandado, Chem. – Eur. J., 2011, 17, 3274–3286 CrossRef CAS PubMed;
(d) P. Pershukevich, I. Shushkevich, E. Makarova and K. Solov'eva, J. Appl. Spectrosc., 2008, 75, 706–713 CrossRef CAS PubMed.
-
(a) A. R. Battersby, Nat. Prod. Rep., 2000, 17, 507–526 RSC;
(b) F.-P. Montforts, B. Gerlach and F. Hoeper, Chem. Rev., 1994, 94, 327–347 CrossRef CAS.
-
(a) B. A. Averill, Chem. Rev., 1996, 96, 2951–2964 CrossRef CAS PubMed;
(b) B. R. Crane, L. M. Siegel and E. D. Getzoff, Science, 1995, 270, 59–67 CAS;
(c) R. J. Krueger and L. M. Siegel, Biochemistry, 1982, 21, 2892–2904 CrossRef CAS;
(d) J. Liu, S. Chakraborty, P. Hosseinzadeh, Y. Yu, S. Tian, I. Petrik, A. Bhagi and Y. Lu, Chem. Rev., 2014, 114, 4366–4469 CrossRef CAS PubMed;
(e)
L. B. Maia and J. J. Moura, Chem. Rev. 20141145273–5357 Search PubMed;
(f) M. J. Murphy, L. M. Siegel, S. R. Tove and H. Kamin, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 612–616 CrossRef CAS;
(g) J. Vega and H. Kamin, J. Biol. Chem., 1977, 252, 896–909 CAS;
(h) L. M. Siegel, M. J. Murphy and H. Kamin, J. Biol. Chem., 1973, 248, 251–264 CAS.
-
(a) A. Eschenmoser, Angew. Chem., Int. Ed. Engl., 1988, 27, 5–39 CrossRef PubMed;
(b) J.-H. Martens, H. Barg, M. Warren and D. Jahn, Appl. Microbiol. Biotechnol., 2002, 58, 275–285 CrossRef CAS PubMed;
(c) E. Raux, C. Thermes, P. Heathcote, A. Rambach and M. J. Warren, J. Bacteriol., 1997, 179, 3202–3212 CAS;
(d) A. I. Scott, Angew. Chem., Int. Ed. Engl., 1993, 32, 1223–1243 CrossRef PubMed;
(e) A. I. Scott, A. J. Irwin, L. M. Siegel and J. Shoolery, J. Am. Chem. Soc., 1978, 100, 7987–7994 CrossRef CAS.
-
(a) A. Battersby, P. Harrison and C. Fookes, J. Chem. Soc., Chem. Commun, 1981, 15, 797–799 Search PubMed;
(b) A. R. Battersby, K. Frobel, F. Hammerschmidt and C. Jones, J. Chem. Soc., Chem. Commun., 1982, 455–457 RSC;
(c) M. H. Block, S. C. Zimmerman, G. B. Henderson, S. P. Turner, S. W. Westwood, F. J. Leeper and A. R. Battersby, J. Chem. Soc., Chem. Commun., 1985, 1061–1063 RSC;
(d) F. P. Montforts, S. Ofner, V. Rasetti, A. Eschenmoser, W. D. Woggon, K. Jones and A. R. Battersby, Angew. Chem., Int. Ed. Engl., 1979, 18, 675–677 CrossRef PubMed;
(e) P. Naab, R. Lattmann, C. Angst and A. Eschenmoser, Angew. Chem., Int. Ed. Engl., 1980, 19, 143–145 CrossRef PubMed.
-
(a) J. M. de Souza, F. F. de Assis, C. Carvalho, J. A. Cavaleiro, T. J. Brocksom and K. T. de Oliveira, Tetrahedron Lett., 2014, 55, 1491–1495 CrossRef CAS PubMed;
(b) M. C. de Souza, L. F. Pedrosa, G. S. Cazagrande, V. F. Ferreira, M. G. Neves and J. A. Cavaleiro, Beilstein J. Org. Chem., 2014, 10, 628–633 CrossRef PubMed;
(c) A. M. Silva, A. C. Tomé, M. G. Neves, A. M. Silva and J. A. Cavaleiro, Chem. Commun., 1999, 1767–1768 RSC;
(d) A. C. Tome, P. S. Lacerda, M. Neves and J. A. Cavaleiro, Chem. Commun., 1997, 1199–1200 RSC;
(e) A. Aggarwal, S. Thompson, S. Singh, B. Newton, A. Moore, R. Gao, X. Gu, S. Mukherjee and C. M. Drain, Photochem. Photobiol., 2014, 90, 419–430 CrossRef CAS PubMed;
(f) S. Singh, A. Aggarwal, S. Thompson, J. P. Tomé, X. Zhu, D. Samaroo, M. Vinodu, R. Gao and C. M. Drain, Bioconjugate Chem., 2010, 21, 2136–2146 CrossRef CAS PubMed;
(g) Y. Harel and J. Manassen, J. Am. Chem. Soc., 1978, 100, 6228–6234 CrossRef CAS;
(h) D. J. Simpson and K. M. Smith, J. Am. Chem. Soc., 1988, 110, 2854–2861 CrossRef CAS;
(i) H. Tamiaki, M. Xu and T. Mizoguchi, Tetrahedron Lett., 2012, 53, 3210–3212 CrossRef CAS PubMed;
(j) H. W. Whitlock Jr., R. Hanauer, M. Oester and B. Bower, J. Am. Chem. Soc., 1969, 91, 7485–7489 CrossRef.
-
(a) Y. Yu, H. Lv, X. Ke, B. Yang and J. L. Zhang, Adv. Synth. Catal., 2012, 354, 3509–3516 CrossRef CAS PubMed;
(b) M. J. Crossley and L. G. King, J. Chem. Soc., Chem. Commun., 1984, 920–922 RSC;
(c) J. R. McCarthy, H. A. Jenkins and C. Bruckner, Org. Lett., 2003, 5, 19–22 CrossRef CAS PubMed;
(d) J. Akhigbe, C. Ryppa, M. Zeller and C. Brückner, J. Org. Chem., 2009, 74, 4927–4933 CrossRef CAS PubMed;
(e) L. Liang, H. B. Lv, Y. Yu, P. Wang and J. L. Zhang, Dalton Trans., 2012, 41, 1457–1460 RSC;
(f) H. B. Lv, B. Y. Yang, J. Jing, Y. Yu, J. Zhang and J. L. Zhang, Dalton Trans., 2012, 41, 3116–3118 RSC;
(g) M. Gouterman, R. J. Hall, G. E. Khalil, P. C. Martin, E. G. Shankland and R. L. Cerny, J. Am. Chem. Soc., 1989, 111, 3702–3707 CrossRef CAS;
(h) C. Bruckner, S. J. Rettig and D. Dolphin, J. Org. Chem., 1998, 63, 2094–2098 CrossRef.
- T. Bruhn and C. Brückner, J. Org. Chem., 2015, 80, 4861–4868 CrossRef CAS PubMed.
-
(a) J. M. Dabrowski, L. G. Arnaut, M. M. Pereira, C. J. P. Monteiro, K. Urbanska, S. Simoes and G. Stochel, ChemMedChem, 2010, 5, 1770–1780 CrossRef CAS PubMed;
(b) M. M. Pereira, C. J. P. Monteiro, A. V. C. Simoes, S. M. A. Pinto, A. R. Abreu, G. F. F. Sa, E. F. F. Silva, L. B. Rocha, J. M. Dabrowski, S. J. Formosinho, S. Simoes and L. G. Arnaut, Tetrahedron, 2010, 66, 9545–9551 CrossRef CAS PubMed;
(c) M. Pineiro, A. Gonsalves, M. M. Pereira, S. J. Formosinho and L. G. Arnaut, J. Phys. Chem. A, 2002, 106, 3787–3795 CrossRef CAS.
-
(a) C. Bruckner and D. Dolphin, Tetrahedron Lett., 1995, 36, 9425–9428 CrossRef;
(b) H. Fischer and H. Eckoldt, Justus Liebigs Ann. Chem., 1940, 544, 138–162 CrossRef PubMed;
(c) C. K. Chang, C. Sotiriou and W. Wu, J. Chem. Soc., Chem. Commun., 1986, 1213–1215 RSC;
(d) Y. H. Chen, C. J. Medforth, K. M. Smith, J. Alderfer, T. J. Dougherty and R. K. Pandey, J. Org. Chem., 2001, 66, 3930–3939 CrossRef CAS PubMed;
(e) J. M. Sutton, N. Fernandez and R. W. Boyle, J. Porphyrins Phthalocyanines, 2000, 4, 655–658 CrossRef CAS.
- A. M. Shulga, I. M. Biteva, I. F. Gurinovich, L. A. Grubina and G. P. Gurinovich, Biofizika, 1977, 22, 771–776 CAS.
-
(a) A. C. Tome, P. S. S. Lacerda, A. M. G. Silva, M. Neves and J. A. S. Cavaleiro, J. Porphyrins Phthalocyanines, 2000, 4, 532–537 CrossRef CAS;
(b) A. C. Tome, P. S. S. Lacerda, M. Neves and J. A. S. Cavaleiro, Chem. Commun., 1997, 13, 1199–1200 RSC.
-
(a) M. C. de Souza, L. F. Pedrosa, G. S. Cazagrande, V. F. Ferreira, M. Neves and J. A. S. Cavaleiro, Beilstein J. Org. Chem., 2014, 10, 628–633 CrossRef PubMed;
(b) J. M. de Souza, F. F. de Assis, C. M. B. Carvalho, J. A. S. Cavaleiro, T. J. Brocksom and K. T. de Oliveira, Tetrahedron Lett., 2014, 55, 1491–1495 CrossRef CAS PubMed;
(c) A. Aggarwal, S. Thompson, S. Singh, B. Newton, A. Moore, R. M. Gao, X. B. Gu, S. Mukherjee and C. M. Drain, Photochem. Photobiol., 2014, 90, 419–430 CrossRef CAS PubMed;
(d) A. M. G. Silva, A. C. Tome, M. Neves, A. M. S. Silva and J. A. S. Cavaleiro, Chem. Commun., 1999, 1767–1768 RSC;
(e) A. M. G. Silva, A. C. Tome, M. Neves, A. M. S. Silva, J. A. S. Cavaleiro, D. Perrone and A. Dondoni, Tetrahedron Lett., 2002, 43, 603–605 CrossRef CAS.
-
(a) J. C. Fitzmaurice, D. J. Williams, P. T. Wood and J. D. Woollins, J. Chem. Soc., Chem. Commun., 1988, 741–743 RSC;
(b) I.
P. Gray, P. Bhattacharyya, A. M. Slawin and J. D. Woollins, Chem. – Eur. J., 2005, 11, 6221–6227 CrossRef CAS PubMed;
(c) M. J. Pilkington, A. M. Slawin, D. J. Williams, P. T. Wood and J. D. Woollins, Heteroat. Chem., 1990, 1, 351–355 CrossRef CAS PubMed;
(d) P. T. Wood and J. D. Woollins, J. Chem. Soc., Chem. Commun., 1988, 1190–1191 RSC.
- M. Mandal, S. Chatterjee and P. Jaisankar, Synlett, 2012, 2615–2618 CAS.
-
(a) Y. Nishiyama, J. Inoue, K. Teranishi, M. Moriwaki and S. Hamanaka, Tetrahedron Lett., 1992, 33, 6347–6350 CrossRef CAS;
(b) Y. Nishiyama, Y. Makino, S. Hamanaka, A. Ogawa and N. Sonoda, Bull. Chem. Soc. Jpn., 1989, 62, 1682–1684 CrossRef CAS.
-
(a) H. Miwa, E. A. Makarova, K. Ishii, E. A. Luk'yanets and N. Kobayashi, Chem. – Eur. J., 2002, 8, 1082–1090 CrossRef CAS;
(b) A. B. J. Parusel and A. Ghosh, J. Phys. Chem. A, 2000, 104, 2504–2507 CrossRef CAS.
- K. M. Barkigia, J. Fajer, L. D. Spaulding and G. J. B. Williams, J. Am. Chem. Soc., 1981, 103, 176–181 CrossRef CAS.
- M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- N. Kobayashi, H. Miwa and V. N. Nemykin, J. Am. Chem. Soc., 2002, 124, 8007–8020 CrossRef CAS PubMed.
-
(a)
C. J. Ballhausen, Introduction to Ligand field Theory, McGraw-Hill, New York, 1962 Search PubMed;
(b)
H. Konami and N. Kobayashi, in The Phthalocyanines-Properties and Applications, ed. A. B. P. L. C. C. Leznoff, VCH, Weinheim, 1996, vol. 4, ch. 9 Search PubMed.
-
(a) J. Michl, J. Am. Chem. Soc., 1978, 100, 6801–6811 CrossRef CAS;
(b) J. Michl, J. Am. Chem. Soc., 1978, 100, 6812–6818 CrossRef CAS;
(c) J. Michl, Pure Appl. Chem., 1980, 52, 1549–1563 CrossRef CAS;
(d)
J. M. N. Kobayashi, Circular Dichroism and Magnetic Circular Dichroism Spectroscopy for Organic Chemists, Royal Society of Chemistry, London, 2011 Search PubMed.
- A. Tajiri and J. Winkler, Z. Naturforsch., A: Phys. Sci., 1983, 38, 1263–1269 Search PubMed.
-
(a) K. Li, J. Pan, S. S. Feng, A. W. Wu, K. Y. Pu, Y. Liu and B. Liu, Adv. Funct. Mater., 2009, 19, 3535–3542 CrossRef CAS PubMed;
(b) Z. Zhang, S. Tongchusak, Y. Mizukami, Y. J. Kang, T. Ioji, M. Touma, B. Reinhold, D. B. Keskin, E. L. Reinherz and T. Sasada, Biomaterials, 2011, 32, 3666–3678 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1047515. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00054h |
|
| This journal is © the Partner Organisations 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Juan
Tang
a,
Zhuo-Yan
Wu
a,
Jia-Zhen
Chen
a,
Nagao
Kobayashi
*b and
Jun-Long
Zhang
*a
b,
Juan
Tang
a,
Zhuo-Yan
Wu
a,
Jia-Zhen
Chen
a,
Nagao
Kobayashi
*b and
Jun-Long
Zhang
*a