
Stable iso-bacteriochlorin mimics from porpholactone: effect of a β-oxazolone moiety on the frontier π-molecular orbitals†

Abstract
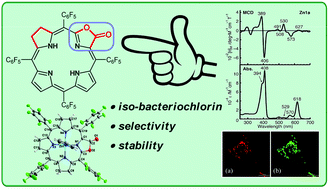
Iso-bacteriochlorins known as siroheme in some reductases, featured with two adjacent reduced pyrrole rings, have distinctive electronic structures from porphyrin, chlorin and bacteriochlorin analogues. However, the synthesis of such cofactor mimics from hydrogenation of chlorin or porphyrin is associated with drawbacks of uncertain regioselectivity and stability. In this work, we present the first example of selective hydrogenation of the adjacent pyrroles in porphyrin or porpholactone free bases assisted by the Woollins reagent (WR). More importantly, adjacent-dihydroporpholactone (1a) displays iso-bacteriochlorin type spectral features and much higher stability under oxidative conditions, compared to the tetrahydroporphyrin analogue (2a). Analysis of magnetic circular dichroism (MCD) spectra and DFT calculations for the frontier π-molecular orbitals for 1a and 2a reveals the significant effect of a β-oxazolone moiety replacement on lowering the HOMO energy level and enhancing the stability resistant to oxidative conditions.
- This article is part of the themed collection: Celebrating 110th Anniversary of Chemistry at Peking University
 Please wait while we load your content...
Please wait while we load your content...

