
Combining coordination and supramolecular chemistry to explore uranyl assembly in the solid state†
Korey P.
Carter
and
Christopher L.
Cahill
*
Department of Chemistry, The George Washington University, 725 21st Street, NW, Washington, D.C. 20052, USA. E-mail: cahill@gwu.edu; Tel: +(202) 994-6959
First published on 10th December 2014
Abstract
The syntheses and crystal structures of twelve new compounds containing the UO22+ cation, a bromo-substituted benzoic acid linker (m-bromo-, p-bromo, or 3,5-dibromobenzoic acid) and a chelating N-donor (1,10-phenanthroline, 2,2′:6′,2′′-terpyridine, or 4′-chloro-2,2′:6′,2′′-terpyridine) are reported. Single crystal X-ray diffraction analyses of these materials allowed for the exploration of the structural relationship between the benzoic acids and the chelating N-donor, as well as the influence of pH on uranyl speciation. At an unadjusted pH (∼3) a mix of uranyl monomers and dimers are observed whereas at higher pH (5–6) uranyl dimers are usually produced with monomers and tetramers also observed. A systematic study of the supramolecular interactions present in these materials was executed by varying the bromine position on the benzoic acid groups along with substituents on the chelating N-donor. Assembly via halogen and hydrogen bonding interactions as well as π–π interactions, including four instances of uranyl oxo-functionalization via halogen bonding, was observed depending on the experimental conditions utilized.
Introduction
Hybrid materials incorporating hexavalent uranium are an area of continued interest due to their penchant for forming structurally diverse coordination polymers and molecular complexes, as well as their relevance to the nuclear fuel cycle.1–9 Development of crystalline uranyl–organic hybrid materials is generally reliant on largely directional metal–ligand interactions to promote extended structures in 1-, 2- and 3-dimensions.10 The resulting structural diversity of uranyl hybrid materials provides a platform for understanding the relationship between solution-phase [UO2]2+ speciation and solid-state manifestations thereof, as is relevant for the delineation of structure property relationships such as luminescence or actinide (An) transport in the environment. The unique chemistry of the linear triatomic [UO2]2+ cation, where additional bonding is generally constrained to the equatorial plane, results in three observed primary building units (square, pentagonal and hexagonal bipyramids). Moreover, hydrolysis of the U(VI) metal center (and subsequent condensation) results in an unpredictable range of secondary building units (dimers, trimers, tetramers, hexamers, sheets, chains, etc.).11,12Uranyl hydrolysis in aqueous solution proceeds viaeqn (1) and governs the formation of oligomeric/polymeric SBUs.
| mUO22+ + nH2O ⇄ (UO2)m(OH)n2m−n + nH+ | (1) |
Metal cation hydrolysis can lead to oligomerization products through the creation of a point-shared hydroxyl group (olation) or thorough a two-step process that results in an oxo bridge (oxolation).13,14 Hydrolysis of the uranyl cation can be influenced by [UO2]2+ concentration and pH with oligomeric species more prevalent at pH values above 4.5.15 Combined with a rich portfolio of ligands with a strong tendency to coordinate to the uranyl cation (e.g. carboxylates and phosphonates) the literature is rich in both SBUs and extended assembly thereof.5,8,16–19 Subsequently, the cumulative effects of both ligand contribution and metal-ion hydrolysis make the synthesis of materials with desired (or at least predictable) topologies rather challenging. While the rich diversity of uranyl materials have proven fruitful for structural characterization, the tuning of the electronic properties of the uranyl ion6,20,21 remains challenging and thus an understanding of how to direct structure–property relationships in uranyl materials remains elusive.4,22,23 As metal-ion hydrolysis prevents predictable construction of uranyl hybrid materials we turn to the molecular solid-state and supramolecular chemistry, as is the focus of this issue.
Supramolecular assembly of materials via attractive noncovalent interactions provides a platform to circumvent hydrolysis related synthetic challenges. A combination of chelating and linking ligands allows for the directed assembly of molecules into crystalline architectures.24 Applications of supramolecular assembly in solid-state materials are broad and continue to expand, yet at present include drug design,25,26 catalysis,27,28 nanomaterials29,30 and organic materials design.31,32 Braga described crystal engineering as “making crystals by design,”33 and by utilizing an understanding of intermolecular interactions in the context of crystal packing and metal–ligand coordination, one can address some of the challenges that stem from a diverse speciation profile. This is a concept that has been explored for transition metal chemistry34 and that has more recently been extended to the actinide series, yet the area remains underexplored.7,35 To take a directed approach to assembly in a U(VI) system, one must find a way to restrict or “shut down” hydrolysis in order to end up with predictable molecular units (or tectons). Previous work in our group has demonstrated that synthesis of [UO2]2+ materials in highly acidic and high halide media will limit uranyl hydrolysis to yield the [UO2X4]2− species (where X = Cl, Br)36–38 or the analogous halide-nitrate ([UO2Cl3NO3]2−)39 almost exclusively, which can then be assembled via the use of supramolecular (hydrogen- and halogen bonding) synthons. More recently, it has been shown that the use of acidic pseudo–halogens (SCN−)40,41 is also quite effective in limiting uranyl speciation and producing anionic discrete building units that can be assembled via a diverse array of supramolecular interactions.
The formation of molecular [UO2]2+ materials does not exclusively require the use of harsh acidic conditions and indeed the literature is rich with a wide variety of uranyl molecular species.16,42–45 This has been highlighted by Forbes et al. in the synthesis of uranyl hybrid materials containing carboxylates and amino acids that can be assembled via hydrogen bonding interactions.46,47 Previous work from our group has also shown that a combination of coordination chemistry principles with a series of halogen functionalized benzoic acids can yield discrete uranyl materials which utilize halogen–halogen interactions for assembly.48 In that study, we relied on pH as a method for controlling uranyl hydrolysis, which while effective therein, remains generally less predictable. An approach to thwarting hydrolysis via coordination chemistry that does not require acidic media may be realized via the use of a chelating ligand to force selection of a single species with an inherent affinity for forming a specific coordination geometry about the [UO2]2+ cation. Due to their preorganization and relatively large binding affinities for f-element ions (specifically the [UO2]2+ cation),49 chelating N-donors such as 1,10-phenanthroline (phen) and 2,2′:6′,2′′-terpyridine (TPY) (and it's derivatives) have been explored as ‘capping’ ligands in the synthesis uranyl coordination polymers,21,50 uranyl molecular materials51 and the formation lanthanide molecular materials that also contain halogen functionalized benzoic acids.52,53
Drawing inspiration from our previous work on uranyl coordination21 and lanthanide supramolecular assembly,52,53 as well as the work of Loiseau and colleagues on uranyl molecular units with TPY,54 we set out to explore a system that features both a halogen functionalized benzoic acid ligand and a chelating N-donor ligand (phen, TPY, Cl-TPY) used for the purpose of controlling hydrolysis and tailoring assembly of the uranyl tectons. Changes in ligand geometry and adjusting pH yield a rich array of molecular tectons containing a diverse array of supramolecular synthons sites (i.e. Br–O halogen bonds, Br–Br and Br–Cl halogen–halogen interactions, Br–π interactions, π–π interactions and hydrogen bonding interactions) that lie at the edge of the immediate coordination sphere. Herein we report the synthesis, crystal structures and modes of supramolecular assembly for a family of twelve new uranyl-bromo benzoic acid-N-donor materials. Additionally, the materials described herein may serve as a platform for delineating uranyl supramolecular assembly criteria based on the observed acceptor–donor pairings. As this is a Frontiers special issue we offer our first of many studies that will explore our motivation to establish a set of comprehensive criteria for supramolecular assembly of actinide species.
Experimental section
Materials and methods
Caution: Whereas the uranium oxyacetate dihydrate (UO2(CH3COO)2·2H2O) used in this study consists of depleted U, standard precautions for handling radioactive and toxic substances should be followed.All materials, including the various bromobenzoic acids (m-bromobenzoic acid, p-bromobenzoic acid and 3,5-dibromobenzoic acid) and chelating N-donors (1,10-phenanthroline, 2,2′:6′,2′′-terpyridine and 4′-chloro-2,2′:6′,2′′-terpyridine) were purchased and used without further purification.
Synthesis
All complexes discussed herein were synthesized via hydrothermal methods at autogeneous pressure in a 23 mL Teflon-lined Parr bomb at an oven temperature of 120 °C for 72 hours. A molar ratio of (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:2:667—UO22+–benzoic acid–phen–water) was used for complexes 1–4, while for complexes 5–12 a molar ratio of (1:2:1:667—UO22+–benzoic acid–terpy–Cl-terpy–water) was optimal for single crystal growth (Table 1). Complexes 4, 8 and 12 could only be produced after the synthesis pH was adjusted via the addition of 25 μL 5 M NaOH. A comprehensive set of synthetic conditions for the UO22+-bromo-benzoic acid-N-donor series are provided in Table 1.
:2:2:667—UO22+–benzoic acid–phen–water) was used for complexes 1–4, while for complexes 5–12 a molar ratio of (1:2:1:667—UO22+–benzoic acid–terpy–Cl-terpy–water) was optimal for single crystal growth (Table 1). Complexes 4, 8 and 12 could only be produced after the synthesis pH was adjusted via the addition of 25 μL 5 M NaOH. A comprehensive set of synthetic conditions for the UO22+-bromo-benzoic acid-N-donor series are provided in Table 1.
| Unadjusted pH | Adjusted pH | |||||
|---|---|---|---|---|---|---|
| m-Bromo | p-Bromo | 3,5-Dibromo | m-Bromo | p-Bromo | 3,5-Dibromo | |
| Phen | 1 | 2 | 3 | 1 | 4 | 3 |
| pHf = 2.5 | pHf = 3.0 | pHf = 2.7 | pHf = 5.2 | pHf = 5.8 | pHf = 5.5 | |
| Terpy | 5 | 6 | 7 | 5 | 8 | 7 |
| pHf = 2.9 | pHf = 2.8 | pHf = 2.9 | pHf = 5.2 | pHf = 5.8 | pHf = 5.6 | |
| Cl-terpy | 9 | 10 | 11 | 9 | 12 | 11 |
| pHf = 2.6 | pHf = 2.8 | pHf = 2.7 | pHf = 5.4 | pHf = 5.5 | pHf = 5.8 | |
Characterization
| 1 | 2 | 3 | 4 | |
|---|---|---|---|---|
| Chem formula | C26H16Br2N2O6U | C26H16Br2N2O6U | C26H16Br4N2O7U | C28H32N4O20U4 |
| Formula weight | 850.26 | 850.26 | 1026.08 | 1696.69 |
| Crystal system | Triclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c | P21/m | P21/n |
| a (Å) | 8.9104(8) | 12.4466(8) | 14.2282(8) | 7.576(6) |
| b (Å) | 12.1555(10) | 21.6789(13) | 6.5817(4) | 15.177(6) |
| c (Å) | 13.0201(11) | 9.4110(6) | 16.1780(9) | 16.860(7) |
| α (°) | 63.614(4) | 90 | 90 | 90 |
| β (°) | 76.124(4) | 95.249(4) | 115.259(5) | 102.094(6) |
| γ (°) | 84.538(3) | 90 | 90 | 90 |
| V (Å3) | 1226.32(19) | 2528.7(3) | 1370.15(15) | 1895.6(19) |
| Z | 2 | 4 | 2 | 2 |
| T (K) | 100 | 293 | 100 | 293 |
| λ (Mo Kα) | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| D calc (g cm−3) | 2.303 | 2.233 | 2.487 | 2.941 |
| μ (mm−1) | 9.923 | 9.624 | 11.805 | 17.118 |
| R int | 0.0375 | 0.0481 | 0.0622 | 0.0296 |
| R 1 [I > 2σ(I)] | 0.0255 | 0.0311 | 0.0279 | 0.0204 |
| wR2 [I > 2σ(I)] | 0.0496 | 0.0623 | 0.0543 | 0.0453 |
| 5 | 6 | 7 | 8 | |
|---|---|---|---|---|
| Chem formula | C36H26Br3N3O12U2 | C26H19Br2N3O6U | C29H17Br4N3O6U | C36H26Br3N3O12U2 |
| Formula weight | 1408.39 | 903.32 | 1061.12 | 1408.39 |
| Crystal system | Triclinic | Triclinic | Monoclinic | Monoclinic |
| Space group |
P |
P |
P21/c | P21/n |
| a (Å) | 8.1393(3) | 8.7005(4) | 13.8471(10) | 20.6384(9) |
| b (Å) | 14.2487(4) | 10.3737(5) | 17.0590(12) | 7.4578(3) |
| c (Å) | 17.1933(5) | 16.6960(8) | 14.0709(10) | 26.5728(12) |
| α (°) | 91.870(3) | 107.848(4) | 90 | 90 |
| β (°) | 97.985(2) | 95.093(4) | 115.488(1) | 105.647(6) |
| γ (°) | 98.728(2) | 95.359(3) | 90 | 90 |
| V (Å3) | 1948.84(11) | 1417.13(12) | 3000.3(4) | 3938.4(3) |
| Z | 2 | 2 | 4 | 4 |
| T (K) | 293 | 293 | 100 | 293 |
| λ (Mo Kα) | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| D calc (g cm−3) | 2.400 | 2.117 | 2.349 | 2.375 |
| μ (mm−1) | 11.441 | 8.595 | 10.785 | 11.322 |
| R int | 0.0389 | 0.0321 | 0.0600 | 0.0671 |
| R 1 [I > 2σ(I)] | 0.0322 | 0.0318 | 0.0273 | 0.0499 |
| wR2 [I > 2σ(I)] | 0.0724 | 0.0695 | 0.0538 | 0.1350 |
| 9 | 10 | 11 | 12 | |
|---|---|---|---|---|
| Chem formula | C36H23Br3N3ClO11U2 | C29H18Br2N3ClO6U | C36H20Br6N3ClO11U2 | C36H25Br3N3ClO12U2 |
| Formula weight | 1424.81 | 937.76 | 1661.52 | 1442.83 |
| Crystal system | Monoclinic | Triclinic | Monoclinic | Monoclinic |
| Space group | P21/n |
P |
P21/c | P21/n |
| a (Å) | 20.7772(8) | 8.9940(3) | 18.8629(13) | 20.7962(8) |
| b (Å) | 7.9807(3) | 10.4251(4) | 8.6591(6) | 7.5283(3) |
| c (Å) | 25.7089(10) | 16.3687(6) | 26.9328(19) | 26.7408(11) |
| α (°) | 90 | 104.094(1) | 90 | 90 |
| β (°) | 110.925(1) | 94.879(1) | 107.231(3) | 106.168(5) |
| γ (°) | 90 | 96.417(2) | 90 | 90 |
| V (Å3) | 3981.8(3) | 1469.19(9) | 4201.6(5) | 4021.0(3) |
| Z | 4 | 2 | 4 | 4 |
| T (K) | 293 | 293 | 100 | 293 |
| λ (Mo Kα) | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| D calc (g cm−3) | 2.377 | 2.120 | 2.627 | 2.383 |
| μ (mm−1) | 11.263 | 8.382 | 13.530 | 11.157 |
| R int | 0.0438 | 0.0468 | 0.0790 | 0.0711 |
| R 1 [I > 2σ(I)] | 0.0300 | 0.0383 | 0.0374 | 0.0390 |
| wR2 [I > 2σ(I)] | 0.0664 | 0.0859 | 0.0715 | 0.0769 |
Description of structures
Single crystal X-ray crystallography analyses revealed three unique building units in this family of molecular complexes: monomers (one unique UO22+ cation) (1–3, 6–7 and 10), dimers (two unique UO22+ cations) (5, 8–9 and 11–12) and a tetramer (two unique UO22+ cations) (4). Local structures are described in detail for complexes 1–6 only as they represent each of the unique observed coordination environments. Modes of supramolecular assembly are described for all complexes however as they are affected by systematic changes in the location of the bromine atoms on the benzoic acid groups and by the nature of the chelating N-donor ligands.Complex 1, [UO2(C12H8N2)(C7H4BrO2)2], crystallizes in the space group P and features an asymmetric unit that contains a uranyl monomer with pentagonal bipyramidal coordination geometry. The [UO2]2+ cation is chelated by a bidentate phen molecule and further coordinated to bidentate and monodentate m-bromobenzoic acid ligands (Fig. 1). U1–O bond distances to the bidentate m-bromobenzoic acid (O3 and O4) are 2.405(2) Å and 2.442(2) Å respectively. The monodentate m-bromobenzoic acid is bound through O5 at a distance of 2.225(3) Å from the uranium center and the bromine atom (Br2) of this ligand facilitates intermolecular Br–O interactions that will be discussed in more detail in the following paragraph. Completing the equatorial coordination sphere of the uranyl ion is the bidentate phen molecule (N1 and N2) and the U1–N distances are 2.552(3) Å and 2.442(2) Å, respectively.
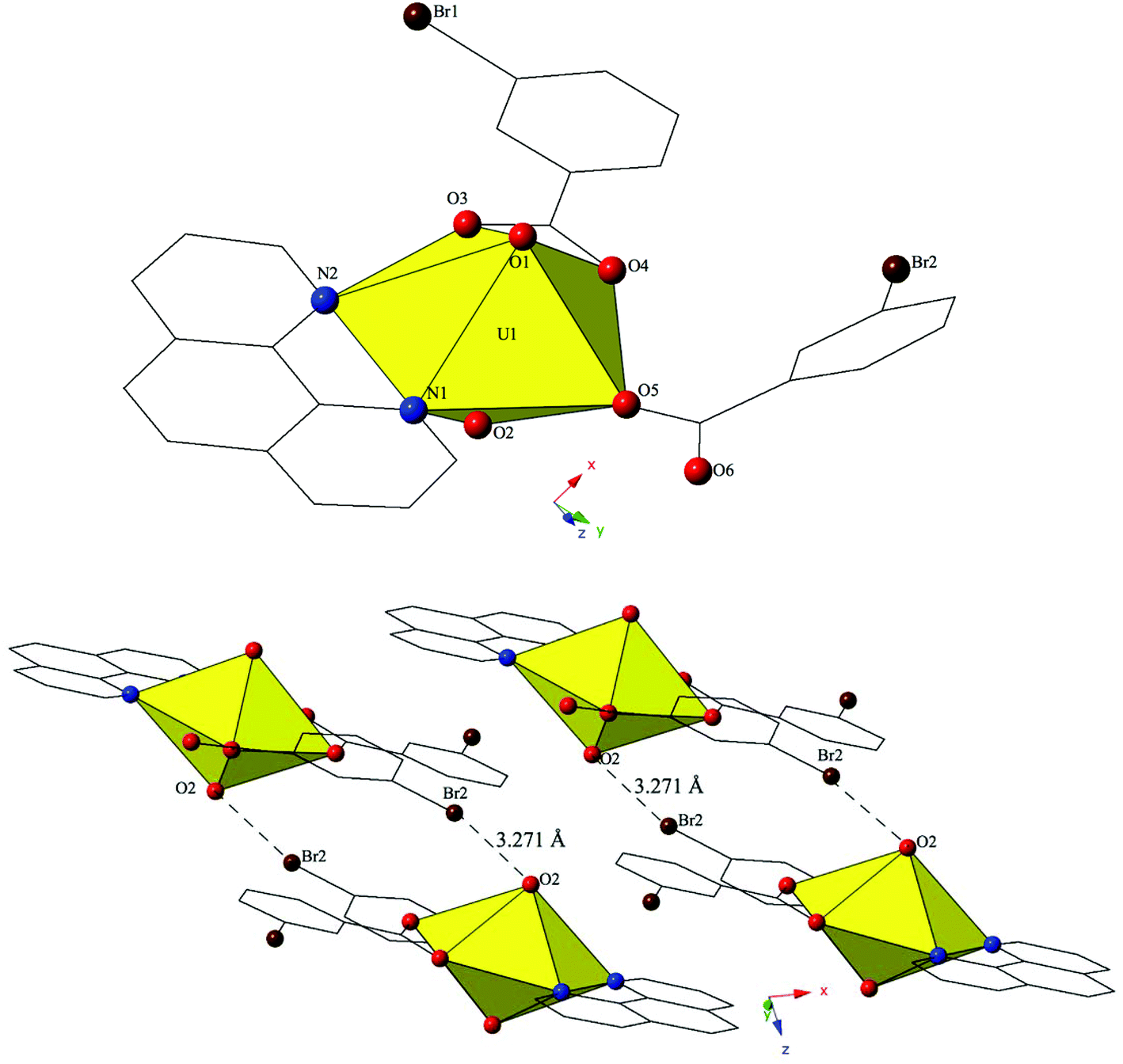 | ||
| Fig. 1 (Top) Polyhedral representation of asymmetric unit of 1. Yellow polyhedra represent uranium metal centers, whereas spheres represent bromine (brown), nitrogen (blue) and oxygen (red). All H atoms have been omitted for clarity. (Bottom) Complex 1 viewed in the (101) plane highlighting the Br–O halogen bonding interactions between uranyl monomers. | ||
The uranyl monomers of 1 are assembled to form molecular dimers via halogen bonds between the axial uranyl oxygen atom (O2) on one unit and the bromine from an m-bromobenzoic acid ligand (Br2) of an adjacent monomer (Fig. 1). The corresponding Br–O interaction distance and angle are 3.271(3) Å and ∠C–Br–O 162.82(14)°. Oxo-functionalization of the uranyl is known in some systems wherein judicious choice of equatorial ligands may enhance Lewis basicity,64–67 yet the uranyl oxygens are typically terminal in most hybrid materials (hence the bipyramidal building units). Some interactions involving the “yl” oxygen atoms are known (i.e. cation–cation interactions (CCIs)),50,68–73 yet these are observed much more frequently for the [NpO2]+ cation.74 Efforts to ‘activate’ the “yl” oxygen atoms in supramolecular chemistry have primarily relied on the “yl” oxygen atoms ability to act as hydrogen bond acceptors.75,76 We have shown that the axial of the uranyl ion can act as a halogen bond acceptor with the [UO2(NCS)4(H2O)]2− anion and the 4-chloropyridine cation41 and in 1 we have the first of four examples where the “yl” oxygen atoms adopts the same function.
The dimers in 1 are assembled into an infinite 2D chain that propagates in the [100] direction via slightly offset π–π stacking interactions77 between phenanthroline ligands on neighboring units. These non-covalent interactions are between the centroid (a calculated centroid, Cg, corresponds to the center of the aromatic ring) of the phen moiety on one unit with the edge of the phen ring on the neighboring unit. Centroids were calculated in the center of the aromatic phen rings participating in these interactions in order to obtain the linear distance (Cg⋯Cg) between the centroids as well as displacement perpendicular to the plane of the phen rings for each of the unique π-stacks (Cg⊥⋯Cg⊥). Additionally, the angle (β) formed by the intersection of the line between centroids and the displacement perpendicular to the plane of the phen rings was determined. As such, the relevant distances and angles for these interactions are: Cg⋯Cg 3.803(2) Å; Cg⊥⋯Cg⊥ 3.3542(14) Å; β = 27.81°.
Changing the position of the bromine from the ortho- to the para-position on the benzoic acid ligand yields complex 2, [UO2(C12H8N2)(C7H4BrO2)2], which crystallizes in the space group P21/c. The asymmetric unit of 2 contains a uranyl monomer with distorted hexagonal bipyramidal coordination geometry. The [UO2]2+ cation is chelated by a bidentate phen molecule along with two bidentate p-bromobenzoic acid ligands (Fig. 2). U1–O bond distances to the two p-bromobenzoic acid ligands (O3, O4, O5 and O6) are at an average distance of 2.466 Å. U1–N distances to the bidentate phen molecule (N1 and N2) are 2.664(4) Å and 2.705 Å respectively and these values are consistent with expected distances for U–N bonds.78,79
 | ||
| Fig. 2 The local coordination geometry of 2 is shown. All H atoms have been omitted for clarity. | ||
Complex 3, [UO2(C12H8N2)(C7H3Br2O2)]·H2O, crystallizes in the space group P21/m and consists of uranyl monomers with a pentagonal bipyramidal coordination geometry. Each [UO2]2+ cation is chelated by a bidentate phen molecule and further coordinated to bidentate and monodentate 3,5-dibromobenzoic acid ligands (Fig. 3). U1–O bond distances to the bidentate 3,5-dibromobenzoic acid (O2 and O3) are each 2.438(4) Å. The monodentate 3,5-dibromobenzoic acid is bound through O4 and is at a distance of 2.235(4) Å from the uranium center. U1–N distances to the bidentate phen molecule (N1 and N2) are 2.561(5) Å and 2.540(5) Å, respectively. The asymmetric unit further contains a lattice water molecule, OW1, which facilitates the supramolecular assembly of the uranyl monomers of 3 into infinite 1D chains.
 | ||
| Fig. 3 (Top) Polyhedral representation of local structure of 3. All H atoms have been omitted for clarity. (Bottom) Complex 3 viewed down the [010] direction. π–π interactions between 3,5-dibromobenzoic acid ligands that assemble 1D chains of 3 are shown. | ||
Offset π–π stacking interactions between 3,5-dibromobenzoic acid ligands on adjacent units link monomers of 3 to form a 1D chain, which propagates infinitely in the [010] direction. The relevant distances and angles for these interactions are Cg⋯Cg 3.5832(14) Å; Cg⊥⋯Cg⊥ 3.2909(10) Å; β = 23.30° (Fig. 3). A bifurcated hydrogen bonding interaction from the lattice water, OW1, occurs with the uranyl axial oxygen atom (O1) and its symmetry equivalent (O1′) and decorates the periphery of the chain (Fig. S1, ESI†). Interaction distances from OW1 to O1 and O1′ are equivalent at 3.234(5) Å.
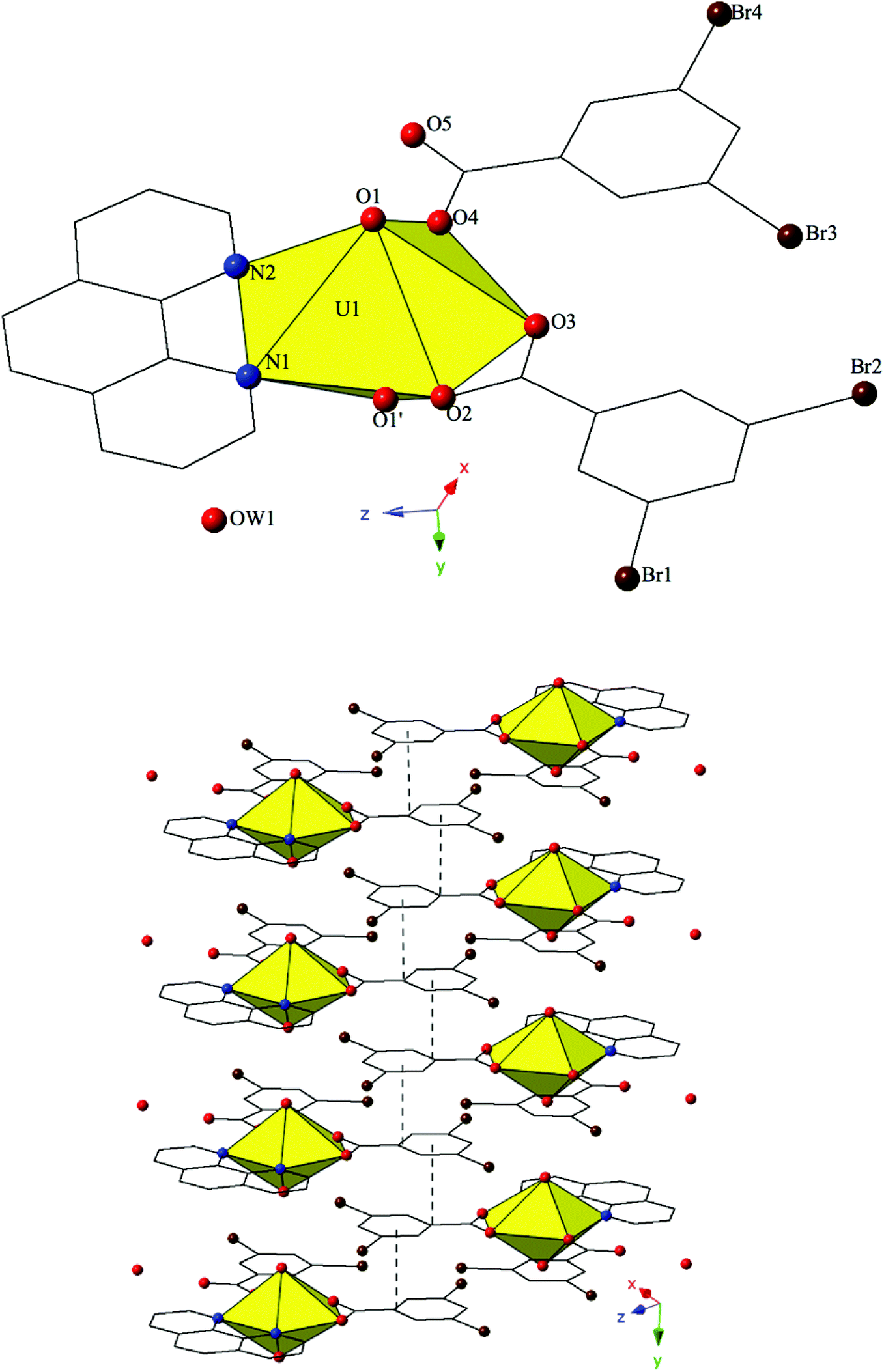
Complex 4, [(UO2)2(OH)(O)(C12H8N2)(CH3COO)(H2O)]2·2H2O, is the first phase described herein to form exclusively at adjusted pH values (approximately 5–6) and crystallizes in the space group P21/c. The asymmetric unit of 4 consists of uranyl tetramer where two unique [UO2]2+ cations, and their symmetry equivalents, have adopted pentagonal bipyramidal coordination geometries (Fig. 4). Both unique uranyl cations are bridged via a point-sharing μ2-OH group (O5) with U–O bond distances of 2.320(3) Å (U1–O5) and 2.355(3) Å (U2–O5) (Confirmed via bond valence calculations, Table S4, ESI†). Oxolation yielded a μ3-O bridge (O6) that connects U1, U2 and U2′ with an average U–O bond distance of 2.264 Å. A third bridging interaction occurs through the coordinated acetate group (O8, O9) and this links U2 with U1′ to complete the tetramer. The corresponding U–O bond distances for the acetate group are 2.359(3) Å (U1′–O9) and 2.376(3) Å (U2–O8), respectively. Bidentate phen groups decorate the periphery of the uranyl tetramer in 4 and complete the equatorial coordination sphere of U1. U1–N distances are 2.673(4) Å (U1–N1) and 2.663(4) Å (U1–N2) which are consistent with the bond lengths observed in complexes 1–3. A bound water molecule (OW1) lies at the apex of pentagonal bipyramid equatorial geometry of U2 at a distance 2.595(4) Å and the asymmetric unit of 4 further contains a lattice water molecule, OW2, which facilitates additional intermolecular interactions that will be discussed below. Despite its inclusion in the initial synthesis, the p-bromobenzoic acid ligand did not incorporate into the final structure of 4 nor was its presence as an impurity detected via PXRD (Fig. S8†).
 | ||
| Fig. 4 (Top) Polyhedral representation of local structure of 4. All H atoms have been omitted for clarity. (Bottom) Complex 4 viewed down approximately the [101] direction. π–π interactions that stitch together the 1D chains of uranyl tetramers are highlighted. | ||
Looking at the global structure of 4, the uranyl tetramers are assembled via offset π–π stacking interactions into a staggered 1D chain that propagates in approximately the [100] direction (Fig. 4). These non-covalent interactions are between the centroid of the phen moiety on one unit with the edge of the phen ring on the neighboring unit. The relevant distances and angles for these interactions are: Cg⋯Cg 3.667(4) Å; Cg⊥⋯Cg⊥ 3.4082(19) Å; β = 21.67°. A bifurcated hydrogen bonding interaction from the lattice water, OW2, occurs with the axial uranyl oxygen atom (O3) of one tetramer and the bound water molecule (OW1) on the tetramer directly below. Interaction distances from OW2 are 2.817(5) Å to O3 and 2.798(6) Å to OW1 and the bifurcated interaction leads to the formation of a 2D sheet in approximately the (101) plane.
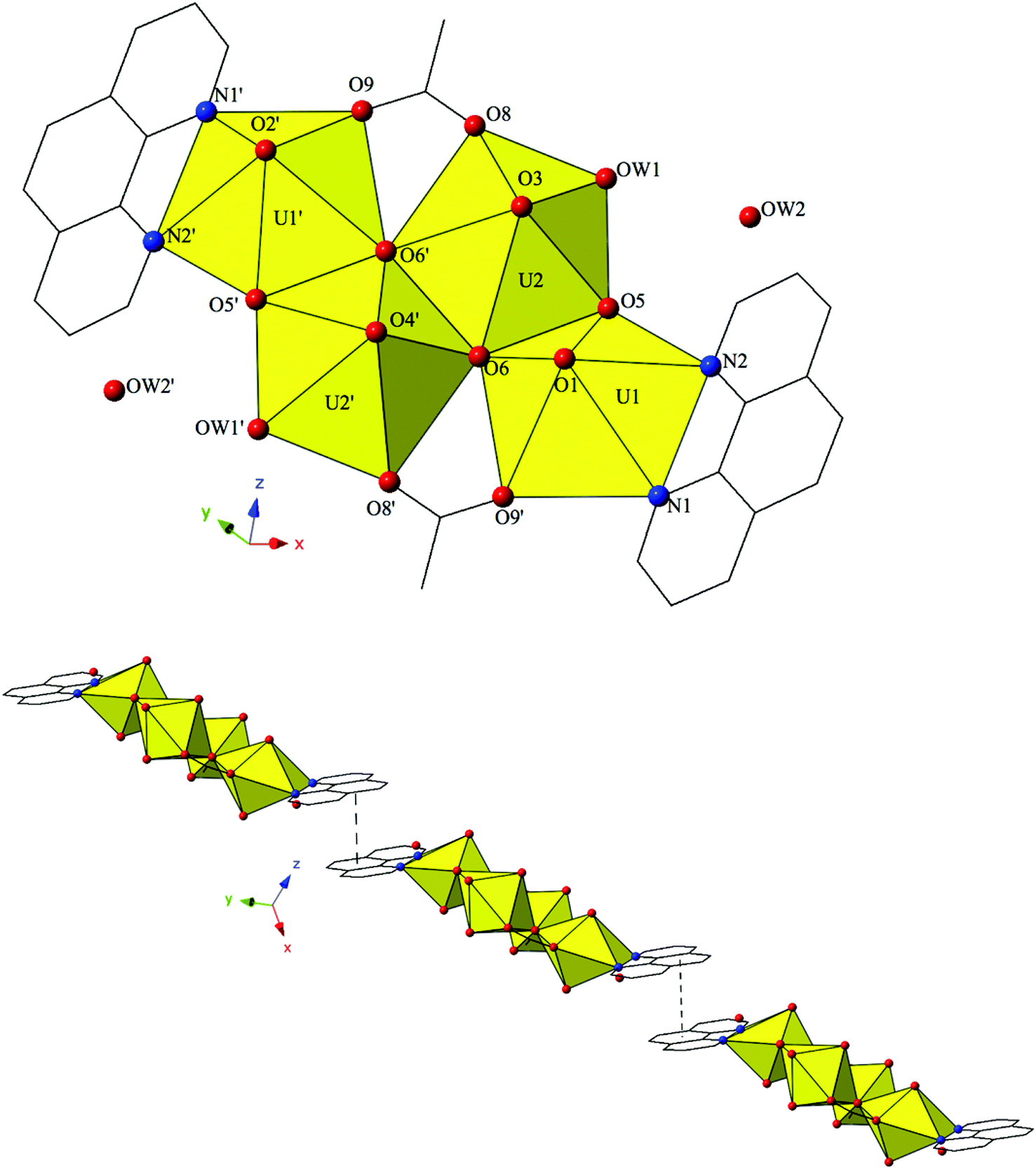
The introduction of terpy as a chelating ligand yields complex 5, [(UO2)2(OH)(C15H11N3)(C7H4BrO2)3]·H2O, which crystallizes in the space group P. The asymmetric unit of 5 consists of a uranyl dimer where both [UO2]2+ cations display pentagonal bipyramidal local coordination geometries (Fig. 5). The two uranyl cations are linked via a bridging bidentate m-bromobenzoic acid ligand (O6 and O7) as well as a point-sharing μ2-OH group (confirmed via bond valence calculations, Table S5, ESI†). Equatorial uranium–oxygen bond distances are 2.398(3) Å (U1–O6) and 2.341(4) Å (U2–O7) for the bridging m-bromobenzoic acid and 2.255(3) Å (U1–O5) and 2.374(3) Å (U2–O5) for the bridging hydroxide. The coordination sphere of U1 is completed by a tridentate terpy molecule bound through its three nitrogen atoms (N1, N2 and N3), with an average U1–N bond distance of 2.587 Å. Two additional m-bromobenzoic acid ligands decorate the U2 metal center and exhibit bidentate and monodentate coordination modes. U2–O bond distances to the bidentate m-bromobenzoic acid ligand (O8 and O9) are 2.453(4) Å and 2.548(3) respectively and the U2–O10 bond distance for the monodentate m-bromobenzoic acid is 2.276(3) Å. A lattice water molecule, OW1, which interacts with carboxylate oxygen (O11) via hydrogen bonding, completes the asymmetric unit.
 | ||
| Fig. 5 (Top) Polyhedral representation of asymmetric unit of 5. All H atoms have been omitted for clarity. (Bottom) Complex 5 viewed down approximately the [011] direction featuring a Type I Br–Br interaction that links the adjacent uranyl dimers. | ||
Looking at the global structure of 5, we see our first example of the halogen–halogen interaction as the uranyl dimers are linked via bromine atoms on adjacent units (Fig. 5). Halogen–halogen interactions tend to adopt one of two geometries in order to minimize the overlap of regions of negative charge density.80,81 The interactions in complex 5 (Br3–Br3′) meet the criteria described by Desiraju et al.82,83 for a Type I halogen–halogen interaction with a distance of 3.4397(13) Å (93.0% vdW) and angles (θ1,θ2) equal to 139.00(19)°. The uranyl dimers of complex 5 propagate as infinite 1D chains via hydrogen bonds in approximately the [100] direction with the lattice water molecule, OW1, interacting with the point sharing hydroxide group, O5, of one dimer directly above and the carboxylate oxygen, O9, of the dimer lying directly below at distances of 2.721(5) Å and 2.808(5) Å, respectively.
Complex 6, [UO2(C15H11N3)(C7H4BrO2)2], crystallizes in the space group P and features an asymmetric unit that contains one pentagonal bipyramidal uranyl PBU. Each [UO2]2+ cation is chelated by a tridentate terpy molecule and coordinated by two monodentate p-bromobenzoic acid ligands (Fig. 6). U1–O bond distances to the monodentate p-bromobenzoic acid ligands (O3 and O5) are 2.230(3) Å and 2.285(3) Å, respectively. The tridentate terpy molecule (N1, N2 and N3) caps the uranyl coordination sphere and the average U1–N distance is 2.582 Å.
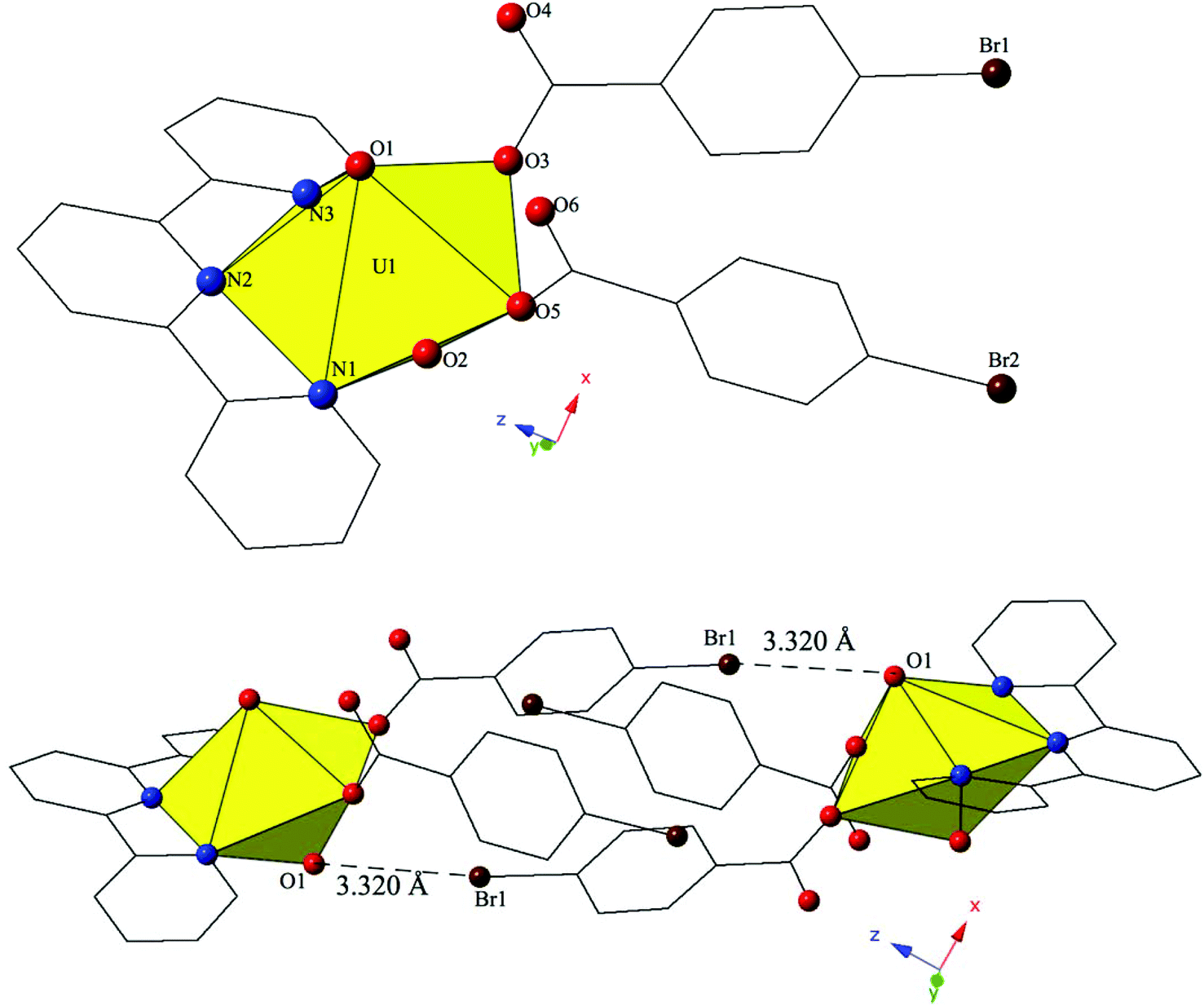 | ||
| Fig. 6 (Top) Polyhedral representation of asymmetric unit of 6. All H atoms have been omitted for clarity. (Bottom) Complex 6 viewed down approximately the [001] direction highlighting Br–O halogen bonding interactions that assemble uranyl monomers into molecular dimers. | ||
The uranyl monomers of 6 form molecular dimers via halogen bonding interactions between the axial uranyl oxygen atom (O1) on one unit and the bromine from a p-bromobenzoic acid ligand (Br1) on the neighboring monomer (Fig. 6). The corresponding Br–O interaction distance and angle are 3.320(3) Å and ∠C–Br–O 144.52(17)°. Additional supramolecular connectivity in 6 is achieved through a series a series of weak hydrogen bonding interactions.
The combination of terpy and the 3,5-dibromobenzoic acid ligand yields complex 7, [UO2(C15H11N3)(C7H3Br2O2)], which crystallizes in the space group P21/c. Complex 7 features nearly identical uranyl coordination geometry to 6, and thus will not be described in detail. The modes of supramolecular assembly appear to be similar when comparing 6 and 7, yet a detailed look reveals significant differences. Similar to 6, uranyl monomers of 7 are tethered to form molecular dimers via halogen bonding interactions between the axial uranyl oxygen atom (O1) on one unit and the bromine from a 3,5-dibromobenzoic acid ligand (Br1) on the neighboring monomer (Fig. 7). The corresponding Br–O interaction distance and angle are 3.246(3) Å and ∠C–Br–O 152.80(13)°. Whereas additional dimensionality in 6 was achieved via weak hydrogen bonding interactions, the supramolecular dimers of 7 are assembled into a zigzag 1D chain via a pair of moderately strong84 localized Br–π (Br2–C7) and (Br4–C3) interactions85 between two 3,5-dibromobenzoic acid ligands on the same uranyl unit with the periphery of the terpyridine moiety on a neighboring unit (Fig. S2, ESI†). Halogen–π interactions are defined as either moderate or strong lone pair–π interactions by Reedijk and colleagues84 based on whether the interaction distance is less than or equal to the corresponding sum of the van der Waals radii (3.550 Å for bromine and carbon). The halogen–π interactions of 7 are at distances of 3.441(4) Å (Br2–C7) and 3.315(4) Å (Br4–C3) respectively and as both of these interactions are well within the sum of the corresponding vdW radii of bromine and carbon this is suggestive that the vdW overlap of the bromine atoms (Br2 and Br4) of the 3,5-dibromobenzoic acid ligands and the aromatic rings of the terpyridine molecules in 7 are significant.
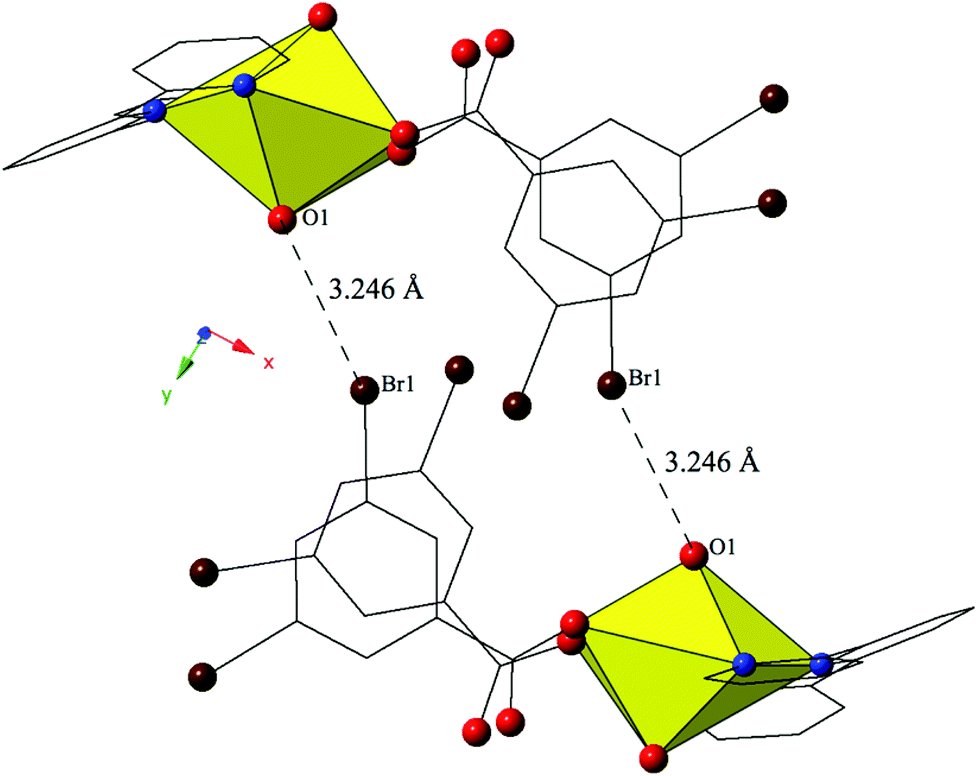 | ||
| Fig. 7 Complex 7 viewed down approximately the [010] direction showing the Br–O halogen bonding interactions that stitch together neighboring uranyl monomers. | ||
Complex 8, [(UO2)2(OH)(C15H11N3)(C7H4BrO2)3], forms at adjusted pH values (5–6) and crystallizes in the space group P21/n. Although the p-bromobenzoic acid ligand is used in the synthesis of 8, the local coordination geometry is similar to 5 and will not be described in detail. The structure of 8 contains uranyl dimers that are linked via localized Br–π interactions, stemming from the bromine of a p-bromobenzoic acid ligand on one unit (Br1) and the periphery of a benzoic acid ring on the neighboring uranyl unit (C31) (Fig. 8). The chains formed from these Br–π interactions propagate in approximately the [101] direction with Br1–C31 interaction distances of 3.315(12) Å. Additionally, a lattice water molecule is absent in 8, which represents a slight variation from the asymmetric unit of 5.
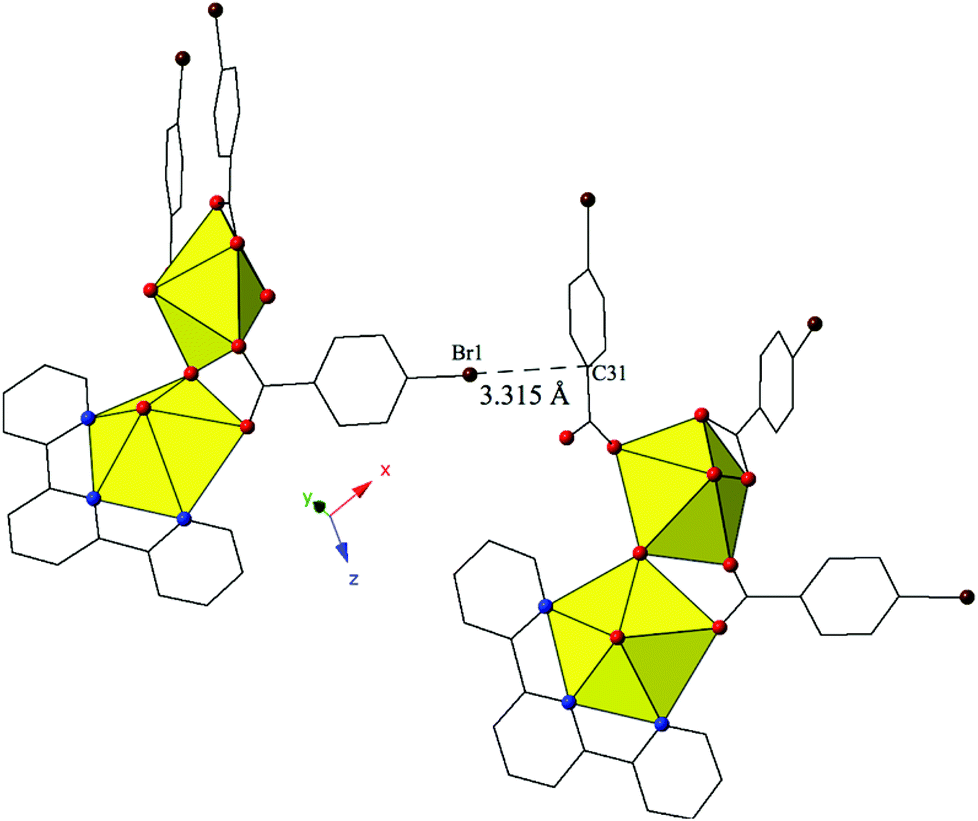 | ||
| Fig. 8 Complex 8 viewed down approximately the [101] direction highlighting the localized Br–π interaction that ties together neighboring uranyl dimers. | ||
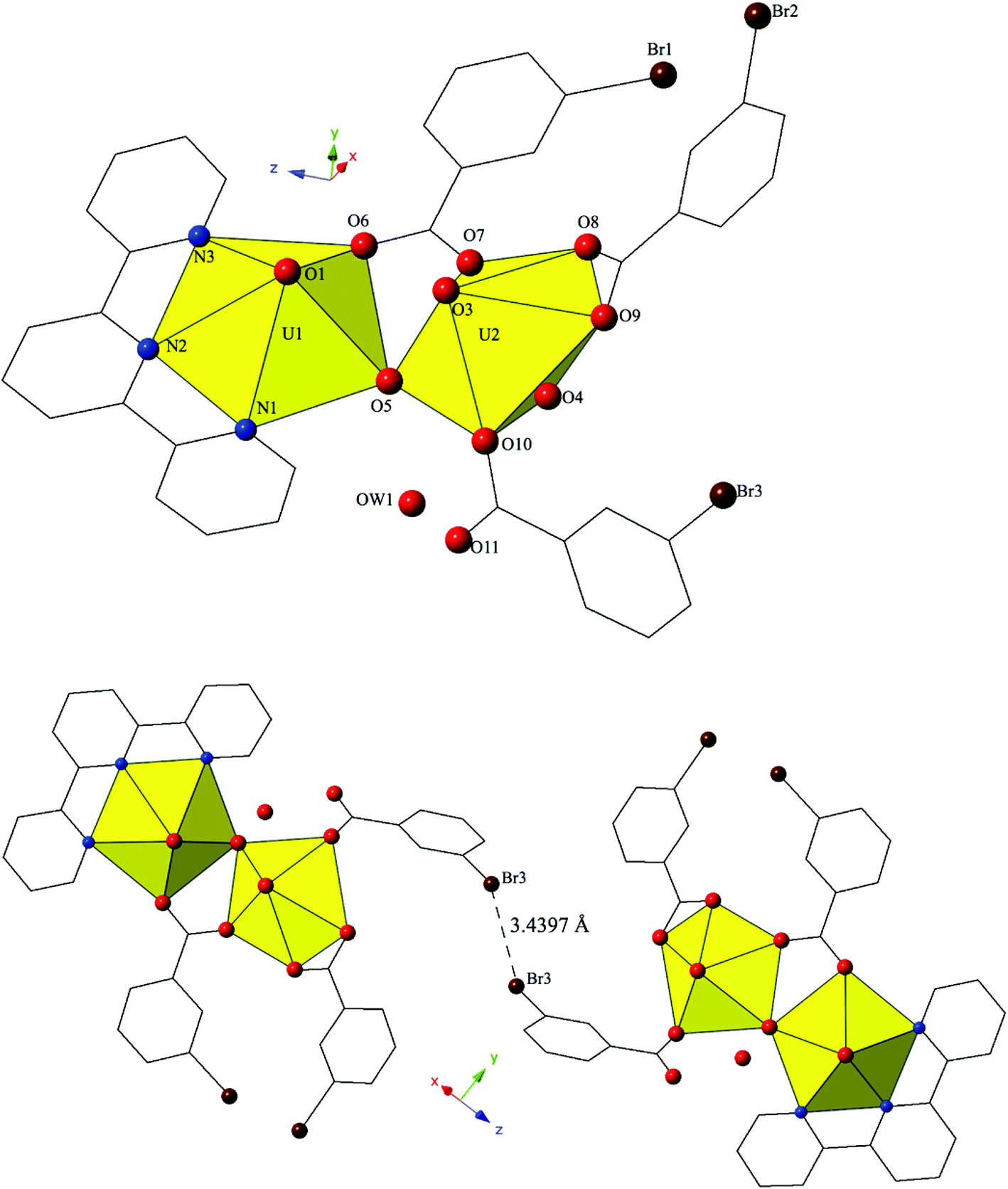
Complex 9, [(UO2)2(OH)(C15H10ClN3)(C7H4BrO2)3], crystallizes in the space group P21/n. Beyond the introduction of Cl-terpy as a capping ligand, 9 features a nearly identical local coordination geometry to 5 and thus will not be described in detail. The only changes from 5 to 9 are the absence of a crystallized lattice water molecule and the addition of a chlorine atom at the 4′-position of the terpy molecule in 9. Whereas these changes may seem insignificant, the resulting supramolecular interactions in 9 have changed dramatically. A bifurcated halogen–halogen interaction (Br3–Cl1 and Br3–Br1) where the bromine atom of one m-bromobenzoic acid ligand (Br3) acts as a halogen bond donor links together the uranyl dimers of 9 into a 1D chain that propagates in the [100] direction (Fig. 9). The bifurcated interaction is made up of a type II83 interaction between the halogen bond donor Br3 and a chlorine (Cl1) at the 4′-position of the terpyridine moiety on an adjacent uranyl unit. The Br3–Cl1 distance is 3.550(2) Å, which is 98.6% of the sum of the van der Waals radii, and the θ1 and θ2 angles are very near ideal type II geometry (θ1 = 180°, θ2 = 90°) at 174.5(2)° and 83.4(2)° respectively. Completing the bifurcated halogen–halogen interaction is a quasi-type I interaction83 between the halogen bond donor Br3 and a bromine (Br1) from an m-bromobenzoic acid ligand on a different neighboring unit. The Br1–Br3 distance is 3.4661(14) Å (93.7% of the sum of the vdW radii) and the θ1 (C19–Br1–Br3) and θ2 (C33–Br3–Br1) values are 163.6(3)° and 148.2(2)°, respectively.
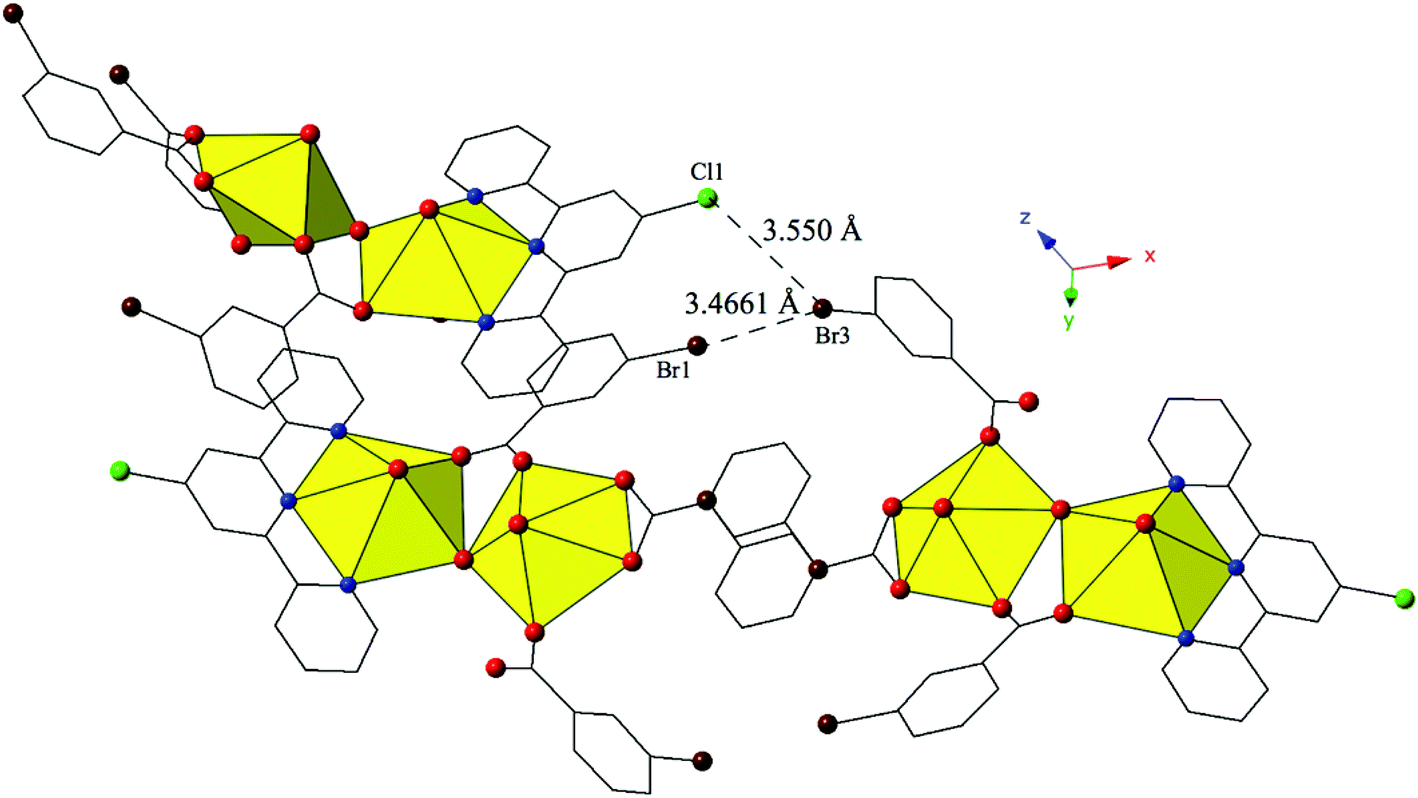 | ||
| Fig. 9 Polyhedral representation of complex 9 viewed down the [111] direction. Green spheres represent chlorine. The bifurcated halogen bonding interaction that connects three neighboring uranyl dimers is highlighted. | ||
Complex 10, [UO2(C15H10ClN3)(C7H4BrO2)2], crystallizes in the space group P and features nearly identical local coordination geometry to 6 and thus will not be described here in detail. The only change from 6 to 10 is the addition of a chlorine atom at the 4′ position of the terpy molecule in 10, which once again results in significant changes in the modes of supramolecular assembly. The uranyl monomers of 10 are assembled into an infinite 1D chain extending approximately along the [011] direction via cooperative Br–O halogen bonding and Br–Cl halogen–halogen interactions (Fig. 10, Fig. S3, ESI†). Halogen bonding interactions between the axial uranyl oxygen of one uranyl monomer (O1) and the bromine of a p-bromobenzoic acid ligand (Br2) on the neighboring unit are similar to those observed in 6 with corresponding interaction distances and angles of 3.319(3) Å and 148.9(2)°, respectively (Fig. 10). In addition, 10 features a type I halogen–halogen interaction83 between the bromine of the p-bromobenzoic acid ligand (Br1) of the uranyl monomer and the chlorine at the 4′-position of the terpy on an adjacent unit. The Br1–Cl1 interaction distance is 3.588(2) Å (99.6% vdW) with a θ1 value of 120.7(2)° and a θ2 value of 118.4(2) (Fig. S3, ESI†).
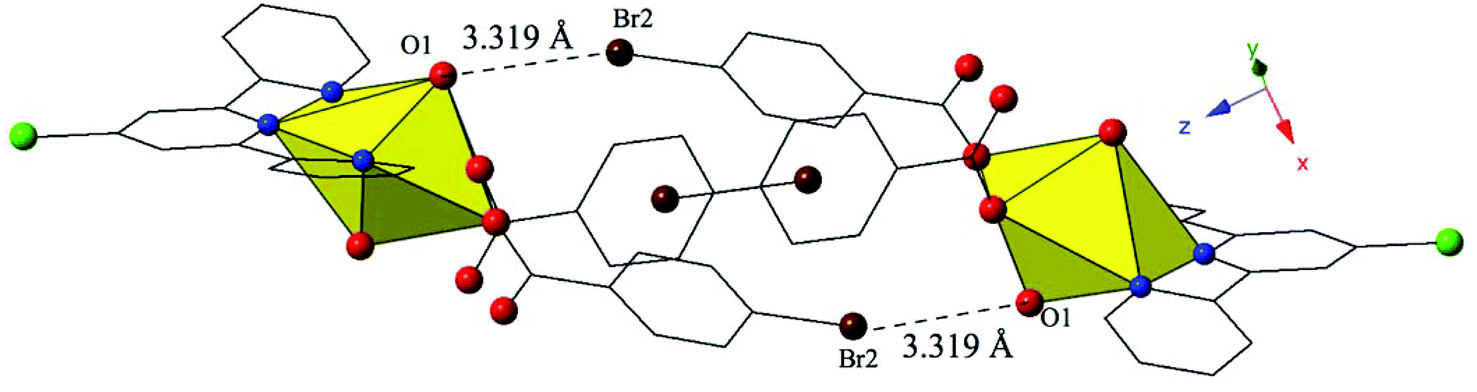 | ||
| Fig. 10 Complex 10 viewed down approximately the [010] direction showing the Br–O halogen bonding interactions that in concert with Br–Cl interactions link neighboring uranyl monomers. | ||
Complex 11, [(UO2)2(OH)(C15H10ClN3)(C7H3Br2O2)3], crystallizes in the space group P21/c and has nearly identical local coordination geometry to 5, so it will not be described here in detail. Similar to 9, 11 lacks a lattice water molecule and features the addition of a chlorine atom at the 4′-position of the terpy molecule as well as the use of 3,5-dibromobenzoic acid ligand in place of the m-bromobenzoic acid ligand used in 5. These changes in ligand geometry yield a global structure that is unlike those observed for complexes 1–10. The major mode of supramolecular assembly is a trifurcated halogen–halogen interaction originating from the chlorine atom at the 4′-position of the TPY molecule, which is acting as both a halogen bond acceptor and donor in 11 (Fig. 11). Generally it is the heavier, more polarizable halogens that behave as halogen bond donors (i.e. iodine)86 but here we observe both a bromine (Br5) and a chlorine (Cl1) atom adopting the role. All three halogen–halogen interactions involving Cl1 (Cl1–Br4, Cl1–Br5 and Cl1–Br6) meet the criteria for type II halogen–halogen interactions (|θ1 − θ2| > 30°) described by Desiraju et al.83 and feature interaction distances of 3.347(2) Å (Cl1–Br4, 93.0% vdW), 3.415(2) Å (Cl1–Br5, 94.9% vdW) and 3.512(2) Å (Cl1–Br6, 97.6% vdW), respectively. Increased connectivity in the structure of 11 is achieved via further supramolecular interactions in the form of a fourth halogen–halogen interaction and a localized Cl–π interaction between Cl1 and the periphery of a 3,5-dibromobenzoic acid ligand on a fourth neighboring unit. The additional halogen–halogen interaction (Br3–Br5) also adopts a type II orientation with an interaction distance of 3.6893(13) Å (99.7 vdW) and θ1 and θ2 values of 150.7(2)° and 68.03(2)°, respectively. The moderate,84 localized Cl–π interaction (Cl1–C20) is at an interaction distance 3.423(7) Å (99.2% of the sum of the vdW radii of chlorine and carbon) and is suggestive of some vdW overlap between the chlorine atom and the benzoic acid ring of the 3,5-dibromobenzoic acid ligand.
 | ||
| Fig. 11 Complex 11 viewed in the (101) plane illustrating the trifurcated halogen–halogen interaction that links together four neighboring uranyl dimers. | ||
Complex 12, [(UO2)2(OH)(C15H10ClN3)(C7H4BrO2)3], is the third phase (along with 4 and 8) that forms only at adjusted pH values (5–6) and crystallizes in the space group P21/n. 12 features nearly identical local coordination geometry to 5 and is isostructural with complex 9, and thus will not be described in detail. Whereas at unadjusted pH with the p-bromobenzoic acid and Cl-TPY ligands we observed a uranyl monomer (10), 12 is a uranyl dimer where the two unique [UO2]2+ cations are bridged by a p-bromobenzoic acid ligand and a point sharing hydroxyl group that is a result of olation.
Similar to 9, 12 features halogen–halogen interactions that assemble the uranyl dimers in approximately the [101] direction (Fig. 12). The interactions between the chlorine at the 4′-position of the TPY molecule (Cl1) and the bromine from the monodentate p-bromobenzoic acid ligand on the neighboring dimer (Br3) can be classified as a type II interaction83 and feature a Cl1–Br3 interaction distance of 3.485(3) Å (96.8% of the sum of the vdW radii) and θ1 and θ2 values of 155.4(5)° and 108.6(3)°, respectively. Further assembly is the result of localized Cl–π interactions between the chlorine at the 4′-position of the TPY molecule and the periphery of an adjacent p-bromobenzoic ligand (C32) (Fig. S4, ESI†). These strong84 Cl–π interactions are at distance of 3.296(9) Å (95.5% vdW) and originate from the same chlorine atoms that are also participating in halogen–halogen interactions described above.
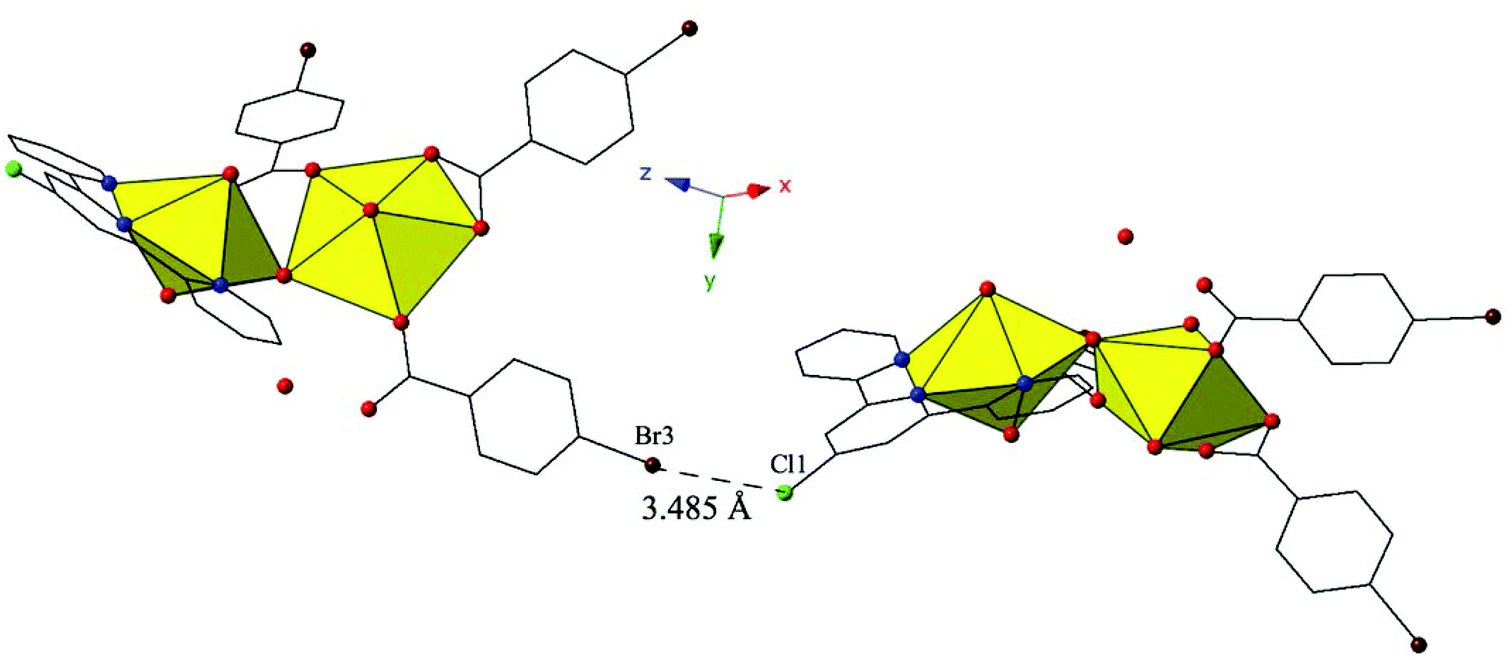 | ||
| Fig. 12 Complex 12 shown in approximately the [101] direction highlighting the Type II Br–Cl interaction that assembles neighboring uranyl dimers. | ||
Structural discussion
As structures 1–12 were synthesized from similar reaction conditions, the resulting structure types and supramolecular synthons provide an opportunity to assess the influence of ligand sterics, the selected chelating N-donor and to a lesser extent, hydrolysis on supramolecular assembly (Table 3).| Complex | Observed synthons | Benzoic acid ligand | Chelating ligand |
|---|---|---|---|
| a m-BrBA = m-bromobenzoic acid, p-BrBA = p-bromobenzoic acid, 3,5-diBrBA = 3,5-dibromobenzoic acid. | |||
| 1 | Br–O | m-BrBA | Phen |
| 2 | N/A | p-BrBA | Phen |
| 3 | H-bonding, π–π | 3,5-diBrBA | Phen |
| 4 | H-bonding, π–π | p-BrBA* (did not incorporate into final structure) | Phen |
| 5 | Br–Br | m-BrBA | Terpy |
| 6 | Br–O | p-BrBA | Terpy |
| 7 | Br–O, Br–π | 3,5-diBrBA | Terpy |
| 8 | Br–π | p-BrBA | Terpy |
| 9 | Br–Br, Br–Cl | m-BrBA | Cl-terpy |
| 10 | Br–Cl, Br–O | p-BrBA | Cl-terpy |
| 11 | Br–Cl (×3), Br–Br, Cl–π | 3,5-diBrBA | Cl-terpy |
| 12 | Br–Cl, Cl–π | p-BrBA | Cl-terpy |
Family 1: complexes with Phen (1–4)
Compounds 1–4 feature a uranyl cation capped by the chelating N-donor phen and further coordinated by bromo-fuctionalized benzoic acid ligands, except in the case of 4 where the p-bromobenzoic acid ligand did not incorporate into the final structure. In 1 and 3 we observe uranyl monomers that adopt pentagonal bipyramidal coordination geometry whereas in the case of 2, we have our only example of a hexagonal bipyramidal coordination geometry. The two unique uranyl cations in the tetramer of 4 also adopt the pentagonal bipyramidal geometries seen in 1 and 3. In this family of phen complexes, supramolecular interactions are observed for complexes 1, 3 and 4, but not complex 2 as hexagonal bipyramidal uranyl geometry does not facilitate additional assembly. These results suggest that uranyl coordination geometry may play some role in supramolecular assembly, yet this was not explored systematically. Complex 1 features uranyl monomers assembled into molecular dimers via halogen bonding interactions between m-bromobenzoic acid ligands and uranyl axial oxygen atoms. In complex 3, where the larger 3,5-dibromobenzoic acid ligand is incorporated, halogens are not involved in supramolecular assembly and the monomers are assembled into 1D chains via a combination of π–π stacking and bifurcated hydrogen bonding interactions. Finally, in complex 4 where the p-bromobenzoic acid ligand did not incorporate we observe assembly of the uranyl tetramers into a 2D sheet via a combination of π–π stacking interactions between phen ligands and bifurcated hydrogen bonding interactions similar to those observed in complex 3.Family 2: complexes with TPY (5–8)
In compounds 5–8, the unique uranyl cations are chelated by the tridentate N-donor terpy and then feature additional coordination to bromo-fuctionalized benzoic acid ligands. Complex 5 is a uranyl dimer that features the m-bromobenzoic acid ligand and is made at both unadjusted (2.5–3) and adjusted pH (5–6). Complexes 6 and 7 both contain uranyl monomers that incorporate the p-bromobenzoic acid and 3,5-dibromobenzoic acid ligands respectively. The former species forms only at unadjusted pH whereas the latter can be made under a range of synthetic conditions. When the pH of the synthesis of 6 is raised to approximately neutral (pH 5–6) the result is complex 8, which is a uranyl dimer that also features the p-bromobenzoic acid ligand. Looking at the modes of assembly of the molecular species in family 2, we see that the dimers of complex 5 are tethered via symmetrical Type I halogen–halogen interactions, the monomers of complexes 6 and 7 utilize halogen bonding interactions to assemble into dimers and 1D chains respectively and the dimers of complex 8 are also stitched into 1D chains via localized Br–π interactions. Br–π interactions, made possible by the additional bromine atoms on the 3,5-dibromobenzoic acid ligands, are also observed in complex 7 and are utilized to achieve additional supramolecular dimensionality.Family 3: complexes with Cl-TPY (9–12)
Compounds 9–12 all feature the chelating Cl-terpy ligand and a bromo-fuctionalized benzoic acid ligand. Complex 10, with the p-bromobenzoic acid ligand, is a monomer made at low (∼3) pH while all other members of this family are uranyl dimers. 9 and 11 are produced at both unadjusted (2.5–3) and adjusted (5–6) pH while 12 is only produced when utilizing the latter conditions. Whereas in families 1 and 2 we observed some variation in the modes of supramolecular assembly, the addition of the chlorine atom at the 4′-position of the terpy molecule in the complexes of family 3 provided the necessary conditions for halogen–halogen interactions to be the dominant mode of assembly. In all four complexes (9–12) we observe at least one unsymmetrical87 (X1 ≠ X2) Cl–Br interaction between molecular units and in complexes 9 and 11 we observe two and three respectively. These interactions, except in complex 10, all adopt type II halogen–halogen geometries,83 which arise from electrophile–nucleophile pairings, and the electrostatic nature of these interactions allows them to be viable at interaction distances near the sum of the vdW radii. In complex 10, the Br–Cl interactions adopt the quasi-type I orientation,83 which is most commonly observed for Cl–Cl contacts,87 yet is not unknown for unsymmetrical Br–Cl interactions.From the results in structural families 1–3 we have noted that the use of the m-bromobenzoic ligand yields assembly via halogen bonding interactions, either halogen–halogen or halogen–heteroatom, independent of the N-donor. Despite its similarities, the p-bromobenzoic acid ligand yields very different results with modes of assembly varying with both the accompanying N-donor and the reaction pH. The sterically larger 3,5-dibromobenzoic acid ligand uses halogen bonding as means of assembly when the N-donor is also of sufficient size (terpy or Cl-terpy). With regards to the chelating N-donors used herein, the bidentate phen was not very useful as crystal engineering tool as it yielded a variety of local coordination modes, yet did not offer much control over the modes of supramolecular assembly. The terpy molecule, however, was able to selectively utilize halogen bonding as means of assembly in all members of family 2, yet there was still some variance in these interactions, whether they were between two halogens, a halogen and a heteroatom or a halogen and a π-system. The Cl-terpy was found to be best tool for crystal engineering the uranyl molecular complexes described herein as all that incorporated the Cl-terpy as a capping ligand were then assembled via unsymmetrical halogen–halogen interactions into extended solid-state structures of varying dimensionalities.
Returning to the participation of the “yl” oxygens in non-covalent interactions, one must be mindful of equatorial ligand contributions towards affecting Lewis basicity.64–67 This, coupled with polarizable halogen donor atoms (such as those observed in 1, 6, 7, and 10), make it difficult to ascertain the specific influence of equatorial ligands versus that of the halogen with respect to “yl” activation. Quantifying these contributions through more systematic syntheses and modeling efforts are ongoing.
Conclusions
The synthesis and crystal structures of twelve uranyl complexes containing bromine functionalized benzoic acids, m-bromobenzoic acid, p-bromobenzoic acid and 3,5-dibromobenzoic acid, and the chelating N-donors phen, terpy and Cl-terpy obtained using hydrothermal reaction conditions have been reported, and their resulting means of supramolecular assembly have been investigated. Throughout the series of structurally diverse materials that were characterized herein, we observe that subtle changes in ligand geometry often lead to significant changes in the interactions utilized for supramolecular assembly. In the materials containing the chelating N-donor Cl-terpy, halogen–halogen interactions were always observed as the functionalization of the back-end of the terpy moiety proved to be a consistent method for the generation of halogen–halogen interactions. In four materials we observed oxo-functionalization of the uranyl via halogen bonding interactions and the ‘activation’ of the uranyl via non-covalent methods is a topic we are continuing to explore. A correlation between the SBU and pH was observed for the materials containing the p-bromobenzoic acid ligand, but not for the m-bromobenzoic acid or 3,5-dibromobenzoic acid ligands. The mechanism for these seemingly divergent results is under investigation. Follow up studies to investigate how changing the character of halogens (more electron withdrawing or more electron donating) on the benzoic acid group can effect the resulting local structures and the corresponding modes for supramolecular assembly. Design of mixed-synthon systems (Br and NO2) and modeling efforts to better understand the role of partial charge in supramolecular interaction strength are also ongoing.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
This material is based upon work supported by the U. S. Department of Energy—Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Sciences, Office of Science, Heavy Elements Program, under grant number DE-FG02-05ER15736.References
- M. Ephritikhine, Dalton Trans., 2006, 2501–2516 RSC.
- C. L. Cahill and L. A. Borkowski, in Structural Chemistry of Inorganic Actinide Compounds, ed. S. V. Krivovichev, P. C. Burns and I. G. Tananaev, Elsevier, Amsterdam, 2007 Search PubMed.
- C. L. Cahill, D. T. de Lill and M. Frisch, CrystEngComm, 2007, 9, 15–26 RSC.
- K.-X. Wang and J.-S. Chen, Acc. Chem. Res., 2011, 44, 531–540 CrossRef CAS PubMed.
- K. E. Knope and C. L. Cahill, in Metal Phosphonate Chemistry: From Synthesis to Applications, ed. A. Clearfield and K. Demadis, Royal Society of Chemistry, London, UK, 2012 Search PubMed.
- L. S. Natrajan, Coord. Chem. Rev., 2012, 256, 1583–1603 CrossRef CAS PubMed.
- M. B. Andrews and C. L. Cahill, Chem. Rev., 2013, 113, 1121–1136 CrossRef CAS PubMed.
- T. Loiseau, I. Mihalcea, N. Henry and C. Volkringer, Coord. Chem. Rev., 2014, 266–267, 69–109 CrossRef CAS PubMed.
- P. Thuéry and J. Harrowfield, Cryst. Growth Des., 2014, 14, 1314–1323 Search PubMed.
- C. Janiak, Dalton Trans., 2003, 2781–2804 RSC.
- C. E. Rowland and C. L. Cahill, Inorg. Chem., 2010, 49, 6716–6724 CrossRef CAS PubMed.
- M. B. Andrews and C. L. Cahill, Angew. Chem., Int. Ed., 2012, 51, 6631–6634 CrossRef CAS PubMed.
- C. F. Baes and R. E. Mesmer, The Hydrolysis of Cations, John Wiley and Sons, New York, NY, 1976 Search PubMed.
- K. E. Knope and L. Soderholm, Chem. Rev., 2013, 113, 944–994 CrossRef CAS PubMed.
- I. Grenthe, J. Fuger, R. J. M. Konings, R. J. Lemire, C. Nguyen-Trun and H. Wanner, Chemical Thermodynamics of Uranium, Organization for Economic Cooperation and Development, Issy-les-Moulineaux, France, 2004 Search PubMed.
- J. Leciejewicz, N. Alcock and T. Kemp, in Coordination Chemistry, Springer, Berlin, Heidelberg, 1995, vol. 82, ch. 2, pp. 43–84 Search PubMed.
- K. E. Knope and C. L. Cahill, Eur. J. Inorg. Chem., 2010, 2010, 1177–1185 CrossRef.
- W. Yang, T. Tian, H.-Y. Wu, Q.-J. Pan, S. Dang and Z.-M. Sun, Inorg. Chem., 2013, 52, 2736–2743 CrossRef CAS PubMed.
- B. Monteiro, J. A. Fernandes, C. C. L. Pereira, S. M. F. Vilela, J. P. C. Tome, J. Marcalo and F. A. Almeida Paz, Acta Crystallogr., Sect. B: Struct. Sci., 2014, 70, 28–36 CAS.
- R. G. Denning, J. Phys. Chem. A, 2007, 111, 4125–4143 CrossRef CAS PubMed.
- S. G. Thangavelu, M. B. Andrews, S. J. A. Pope and C. L. Cahill, Inorg. Chem., 2013, 52, 2060–2069 CrossRef CAS PubMed.
- G. Liu, N. P. Deifel, C. L. Cahill, V. V. Zhurov and A. A. Pinkerton, J. Phys. Chem. A, 2012, 116, 855–864 CrossRef CAS PubMed.
- G. Liu, L. Rao and G. Tian, Phys. Chem. Chem. Phys., 2013, 15, 17487–17495 RSC.
- C. B. Aakeroy, P. D. Chopade, C. Ganser and J. Desper, Chem. Commun., 2011, 47, 4688–4690 RSC.
- Y. Lu, T. Shi, Y. Wang, H. Yang, X. Yan, X. Luo, H. Jiang and W. Zhu, J. Med. Chem., 2009, 52, 2854–2862 CrossRef CAS PubMed.
- R. Wilcken, M. O. Zimmermann, A. Lange, A. C. Joerger and F. M. Boeckler, J. Med. Chem., 2012, 56, 1363–1388 CrossRef PubMed.
- R. R. Knowles and E. N. Jacobsen, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20678–20685 CrossRef CAS PubMed.
- W. Tang, S. Johnston, J. A. Iggo, N. G. Berry, M. Phelan, L. Lian, J. Bacsa and J. Xiao, Angew. Chem., Int. Ed., 2013, 52, 1668–1672 CrossRef CAS PubMed.
- P. H. Dinolfo and J. T. Hupp, Chem. Mater., 2001, 13, 3113–3125 CrossRef CAS.
- T. Kudernac, S. Lei, J. A. A. W. Elemans and S. De Feyter, Chem. Soc. Rev., 2009, 38, 402–421 RSC.
- S. I. Stupp and L. C. Palmer, Chem. Mater., 2014, 26, 507–518 CrossRef CAS.
- D. Wang, G. Tong, R. Dong, Y. Zhou, J. Shen and X. Zhu, Chem. Commun., 2014, 50, 11994–12017 RSC.
- D. Braga, Chem. Commun., 2003, 2751–2754 RSC.
- Y. E. Alexeev, B. I. Kharisov, T. C. H. García and A. D. Garnovskii, Coord. Chem. Rev., 2010, 254, 794–831 CrossRef CAS PubMed.
- R. J. Baker, Chem. – Eur. J., 2012, 18, 16258–16271 CrossRef CAS PubMed.
- N. P. Deifel and C. L. Cahill, CrystEngComm, 2009, 11, 2739–2744 RSC.
- N. P. Deifel and C. L. Cahill, C. R. Chim., 2010, 13, 747–754 CrossRef CAS PubMed.
- M. B. Andrews and C. L. Cahill, Dalton Trans., 2012, 41, 3911–3914 RSC.
- M. B. Andrews and C. L. Cahill, CrystEngComm, 2013, 15, 3082–3086 RSC.
- C. E. Rowland, M. G. Kanatzidis and L. Soderholm, Inorg. Chem., 2012, 51, 11798–11804 CrossRef CAS PubMed.
- R. G. Surbella III and C. L. Cahill, CrystEngComm, 2014, 16, 2352–2364 RSC.
- N. W. Alcock, D. J. Flanders and D. Brown, J. Chem. Soc., Dalton Trans., 1985, 1001–1007 RSC.
- J.-C. Berthet, M. Nierlich and M. Ephritikhine, Dalton Trans., 2004, 2814–2821 RSC.
- P. Thuéry, Inorg. Chem., 2012, 52, 435–447 CrossRef PubMed.
- X.-S. Zhai, Y.-Q. Zheng, J.-L. Lin and W. Xu, Inorg. Chim. Acta, 2014, 423(Part A), 1–10 CrossRef CAS PubMed.
- D. K. Unruh, K. Gojdas, E. Flores, A. Libo and T. Z. Forbes, Inorg. Chem., 2013, 52, 10191–10198 CrossRef CAS PubMed.
- J. de Groot, K. Gojdas, D. K. Unruh and T. Z. Forbes, Cryst. Growth Des., 2014, 14, 1357–1365 CAS.
- N. P. Deifel and C. L. Cahill, Chem. Commun., 2011, 47, 6114–6116 RSC.
- R. D. Hancock, Chem. Soc. Rev., 2013, 42, 1500–1524 RSC.
- P. O. Adelani and P. C. Burns, Inorg. Chem., 2012, 51, 11177–11183 CrossRef CAS PubMed.
- P. Thuéry, Eur. J. Inorg. Chem., 2013, 2013, 4563–4573 CrossRef.
- K. P. Carter, S. J. A. Pope and C. L. Cahill, CrystEngComm, 2014, 16, 1873–1884 RSC.
- K. P. Carter, C. H. F. Zulato and C. L. Cahill, CrystEngComm, 2014, 16, 10189–10202 RSC.
- J. Lhoste, N. Henry, T. Loiseau, Y. Guyot and F. Abraham, Polyhedron, 2013, 50, 321–327 CrossRef CAS PubMed.
- SAINT, Bruker AXS Inc., Madison, Wisconsin, USA, 2007 Search PubMed.
- APEX2, Bruker AXS Inc., Madison, Wisconsin, USA, 2008 Search PubMed.
- SADABS, Bruker AXS, Madison, Wisconsin, USA, 2008 Search PubMed.
- TWINABS, Bruker AXS, Madison, Wisconsin, USA, 2008 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435–435 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- Crystal Maker, Crystal Maker Software Limited, Bicester, England, 2009 Search PubMed.
- JADE, Materials Data Inc., Livermore, California, USA, 2003 Search PubMed.
- M. J. Sarsfield and M. Helliwell, J. Am. Chem. Soc., 2004, 126, 1036–1037 CrossRef CAS PubMed.
- S. Fortier and T. W. Hayton, Coord. Chem. Rev., 2010, 254, 197–214 CrossRef CAS PubMed.
- P. L. Arnold, A.-F. Pécharman, E. Hollis, A. Yahia, L. Maron, S. Parsons and J. B. Love, Nat. Chem., 2010, 2, 1056–1061 CrossRef CAS PubMed.
- A. J. Lewis, H. Yin, P. J. Carroll and E. J. Schelter, Dalton Trans., 2014, 43, 10844–10851 RSC.
- P. Thuéry, M. Nierlich, B. Souley, Z. Asfari and J. Vicens, J. Chem. Soc., Dalton Trans., 1999, 2589–2594 RSC.
- Y. Li, C. L. Cahill and P. C. Burns, Chem. Mater., 2001, 13, 4026–4031 CrossRef CAS.
- K.-A. Kubatko and P. C. Burns, Inorg. Chem., 2006, 45, 10277–10281 CrossRef CAS PubMed.
- J. Lhoste, N. Henry, P. Roussel, T. Loiseau and F. Abraham, Dalton Trans., 2011, 40, 2422–2424 RSC.
- R. C. Severance, M. D. Smith and H.-C. zur Loye, Inorg. Chem., 2011, 50, 7931–7933 CrossRef CAS PubMed.
- V. N. Serezhkin, G. V. Sidorenko, D. V. Pushkin and L. B. Serezhkina, Radiochemistry, 2014, 56, 115–133 CrossRef CAS.
- N. N. Krot and M. S. Grigoriev, Russ. Chem. Rev., 2004, 73, 89–100 CrossRef CAS PubMed.
- D. L. Clark, S. D. Conradson, R. J. Donohoe, D. W. Keogh, D. E. Morris, P. D. Palmer, R. D. Rogers and C. D. Tait, Inorg. Chem., 1999, 38, 1456–1466 CrossRef CAS.
- L. A. Watson and B. P. Hay, Inorg. Chem., 2011, 50, 2599–2605 CrossRef CAS PubMed.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- N. W. Alcock, D. J. Flanders, M. Pennington and D. Brown, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 247–250 CrossRef.
- J.-C. Berthet, M. Nierlich and M. Ephritikhine, Chem. Commun., 2003, 1660–1661 RSC.
- F. F. Awwadi, R. D. Willett, K. A. Peterson and B. Twamley, Chem. – Eur. J., 2006, 12, 8952–8960 CrossRef CAS PubMed.
- L. Brammer, G. Minguez Espallargas and S. Libri, CrystEngComm, 2008, 10, 1712–1727 RSC.
- G. R. Desiraju and R. Parthasarathy, J. Am. Chem. Soc., 1989, 111, 8725–8726 CrossRef CAS.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514–2524 CrossRef CAS PubMed.
- T. J. Mooibroek, P. Gamez and J. Reedijk, CrystEngComm, 2008, 10, 1501–1515 RSC.
- D. Schollmeyer, O. V. Shishkin, T. Ruhl and M. O. Vysotsky, CrystEngComm, 2008, 10, 715–723 RSC.
- P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Chem. Commun., 2008, 1635–1637 RSC.
- S. Tothadi, S. Joseph and G. R. Desiraju, Cryst. Growth Des., 2013, 13, 3242–3254 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic files in CIF format, ORTEP figures of all compounds, PXRD spectra of all compounds, tables of selected supramolecular interaction distances and bond lengths, additional figures for complexes 3, 7, 10 and 12 and bond valence calculations are all available. CCDC 1025739–1025750 for compounds 1–12. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4qi00183d |
| This journal is © the Partner Organisations 2015 |